

mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells
- PMID: 23538752
- PMCID: PMC4225699
- DOI: 10.1038/leu.2013.93
mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells
Abstract
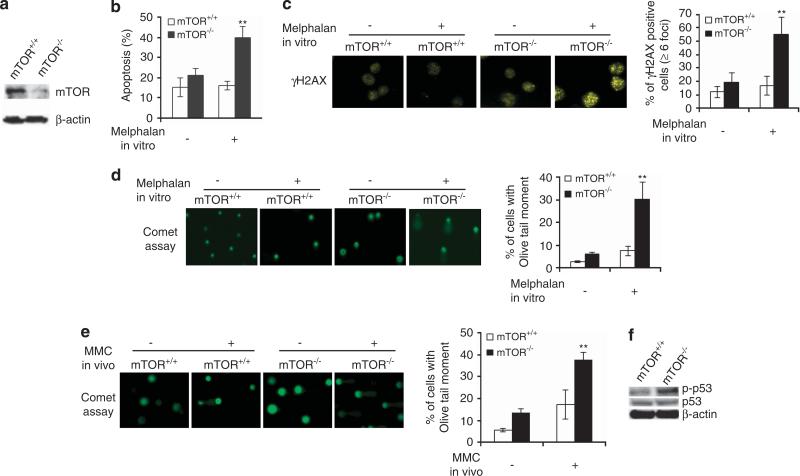
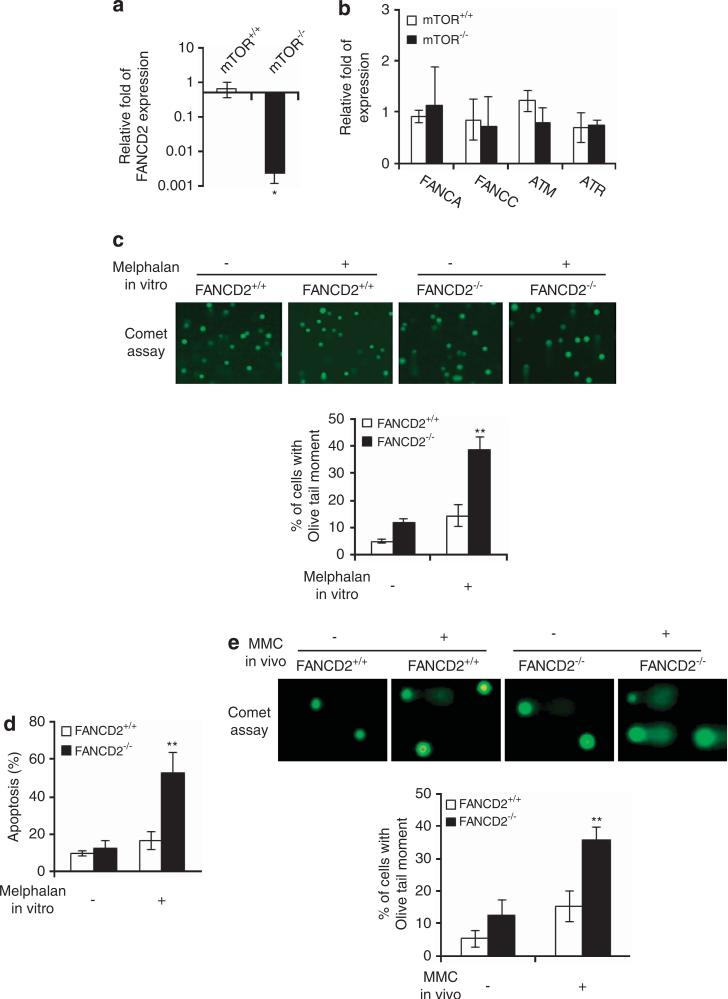
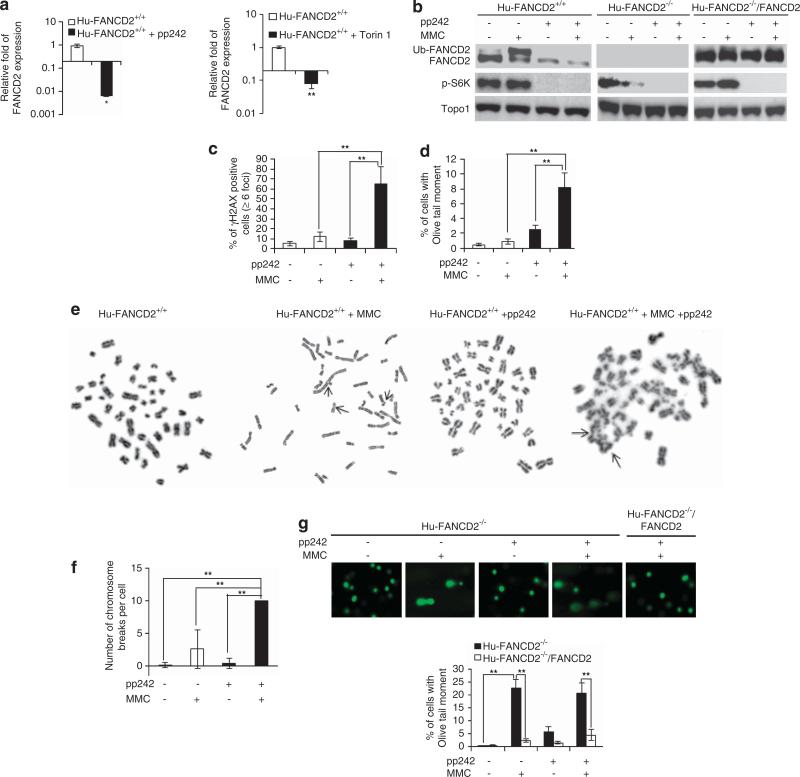
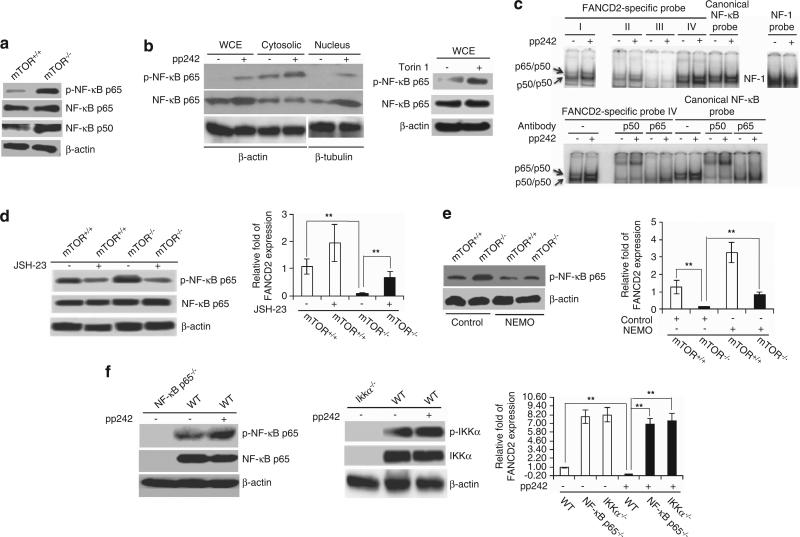
Hematopoietic stem/progenitor cells (HSPCs) function to give rise to mature blood cells. Effective DNA damage response (DDR) and maintenance of genomic stability are crucial for normal functioning of HSPCs. Mammalian target of rapamycin (mTOR) integrates signals from nutrients and growth factors to control protein synthesis, cell growth, survival and metabolism, and has been shown to regulate DDR in yeast and human cancer cells through the p53/p21 signaling cascade. Here, we show that gene targeting of mTOR in HSPCs causes a defective DDR due to a variety of DNA damage agents, mimicking that caused by deficient FANCD2, a key component of the Fanconi anemia (FA) DDR machinery. Mechanistically, mTOR(-/-) HSPCs express drastically reduced FANCD2. Consistent with these genetic findings, inactivation of mTOR in human lymphoblast cells by pp242 or Torin 1, mTOR kinase inhibitors, suppresses FANCD2 expression and causes a defective DDR that can be rescued by reconstitution of exogenous FANCD2. Further mechanistic studies show that mTOR deficiency or inactivation increases phosphorylation and nuclear translocation of nuclear factor (NF)-κB, which results in an enhanced NF-κB binding to FANCD2 promoter to suppress FANCD2 expression. Thus, mTOR regulates DDR and genomic stability in hematopoietic cells through a noncanonical pathway involving NF-κB-mediated FANCD2 expression.
Figures
Similar articles
-
Radiologic differences between bone marrow stromal and hematopoietic progenitor cell lines from Fanconi Anemia (Fancd2(-/-)) mice.Radiat Res. 2014 Jan;181(1):76-89. doi: 10.1667/RR13405.1. Epub 2014 Jan 7. Radiat Res. 2014. PMID: 24397476 Free PMC article.
-
Forkhead transcription factor FoxF1 interacts with Fanconi anemia protein complexes to promote DNA damage response.Oncotarget. 2016 Jan 12;7(2):1912-26. doi: 10.18632/oncotarget.6422. Oncotarget. 2016. PMID: 26625197 Free PMC article.
-
Genomic instability in mice is greater in Fanconi anemia caused by deficiency of Fancd2 than Fancg.Cancer Res. 2010 Dec 1;70(23):9703-10. doi: 10.1158/0008-5472.CAN-09-1022. Epub 2010 Nov 30. Cancer Res. 2010. PMID: 21118969 Free PMC article.
-
Endogenous DNA Damage Leads to p53-Independent Deficits in Replicative Fitness in Fetal Murine Fancd2-/- Hematopoietic Stem and Progenitor Cells.Stem Cell Reports. 2016 Nov 8;7(5):840-853. doi: 10.1016/j.stemcr.2016.09.005. Epub 2016 Oct 6. Stem Cell Reports. 2016. PMID: 27720904 Free PMC article.
-
Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks.Cancer Res. 2013 Jun 1;73(11):3393-401. doi: 10.1158/0008-5472.CAN-12-4282. Epub 2013 Apr 30. Cancer Res. 2013. PMID: 23633493 Free PMC article.
Cited by
-
Fanconi anemia and mTOR pathways functionally interact during stalled replication fork recovery.FEBS Lett. 2021 Mar;595(5):595-603. doi: 10.1002/1873-3468.14035. Epub 2021 Jan 28. FEBS Lett. 2021. PMID: 33423298 Free PMC article.
-
Inhibition of MEK confers hypersensitivity to X-radiation in the context of BRAF mutation in a model of childhood astrocytoma.Pediatr Blood Cancer. 2015 Oct;62(10):1768-74. doi: 10.1002/pbc.25579. Epub 2015 May 15. Pediatr Blood Cancer. 2015. PMID: 25981859 Free PMC article.
-
Pharmacologic Induction of BRCAness in BRCA-Proficient Cancers: Expanding PARP Inhibitor Use.Cancers (Basel). 2022 May 26;14(11):2640. doi: 10.3390/cancers14112640. Cancers (Basel). 2022. PMID: 35681619 Free PMC article. Review.
-
The Intra- and Extra-Telomeric Role of TRF2 in the DNA Damage Response.Int J Mol Sci. 2021 Sep 14;22(18):9900. doi: 10.3390/ijms22189900. Int J Mol Sci. 2021. PMID: 34576063 Free PMC article. Review.
-
A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A.Metabolites. 2021 Dec 21;12(1):6. doi: 10.3390/metabo12010006. Metabolites. 2021. PMID: 35050128 Free PMC article.
References
-
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. - PubMed
-
- Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. - PubMed
-
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. - PubMed
-
- Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, et al. A distinctive DNA damage response in human hematopoietic stem cell reveals an apoptosis-independent role of p53 in self-renewal. Cell Stem Cell. 2010;7:186–197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
