

Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity
- PMID: 23525077
- PMCID: PMC3678719
- DOI: 10.1038/ng.2591
Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity
Abstract
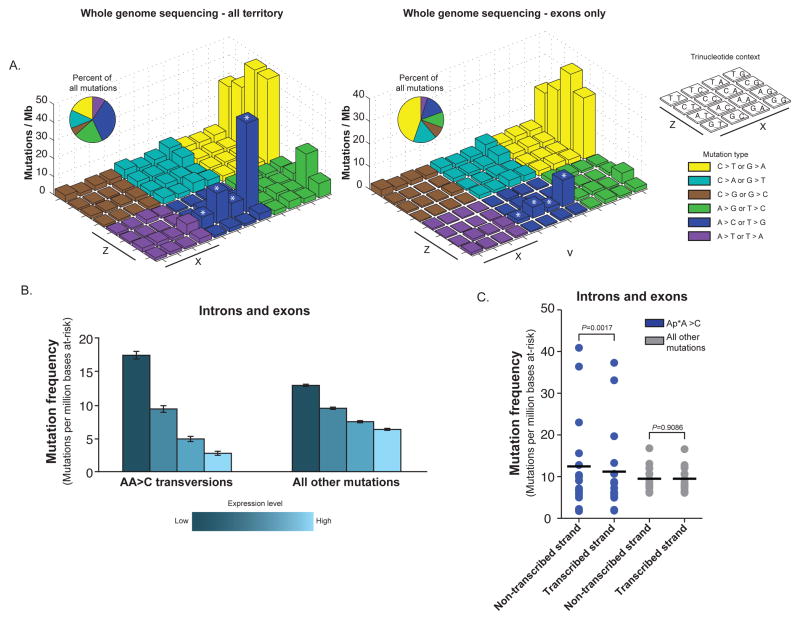
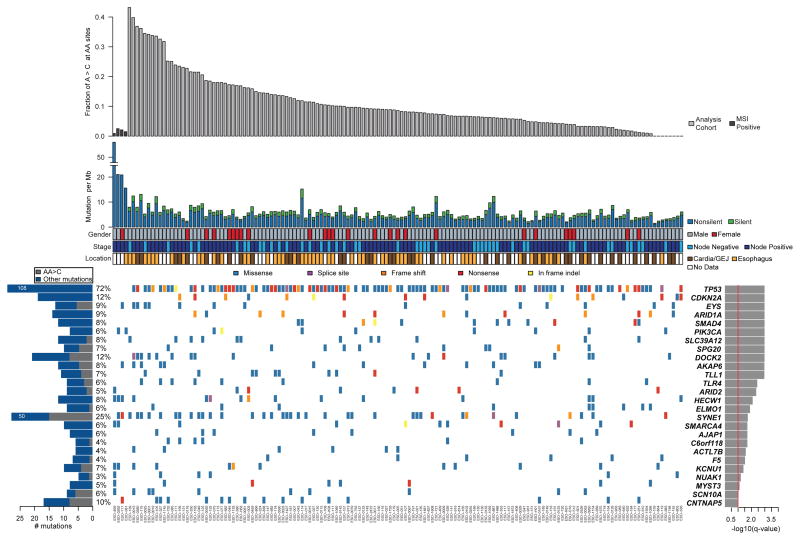
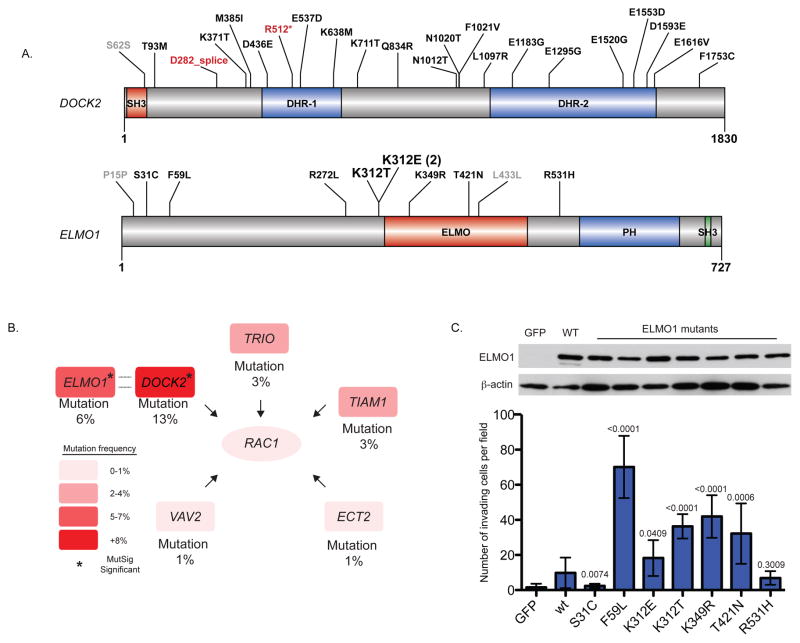
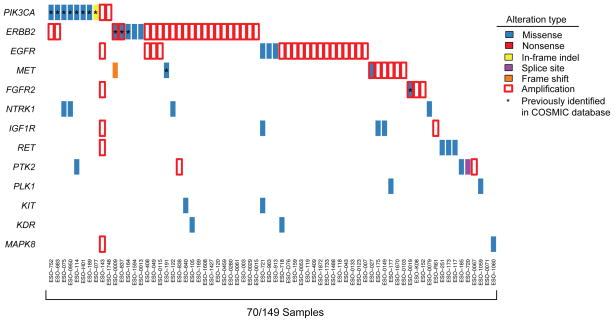
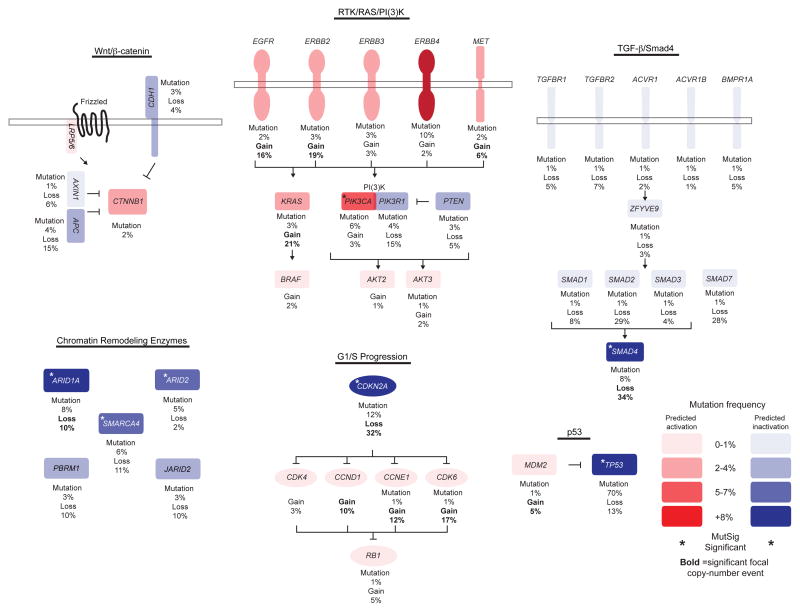
The incidence of esophageal adenocarcinoma (EAC) has risen 600% over the last 30 years. With a 5-year survival rate of ~15%, the identification of new therapeutic targets for EAC is greatly important. We analyze the mutation spectra from whole-exome sequencing of 149 EAC tumor-normal pairs, 15 of which have also been subjected to whole-genome sequencing. We identify a mutational signature defined by a high prevalence of A>C transversions at AA dinucleotides. Statistical analysis of exome data identified 26 significantly mutated genes. Of these genes, five (TP53, CDKN2A, SMAD4, ARID1A and PIK3CA) have previously been implicated in EAC. The new significantly mutated genes include chromatin-modifying factors and candidate contributors SPG20, TLR4, ELMO1 and DOCK2. Functional analyses of EAC-derived mutations in ELMO1 identifies increased cellular invasion. Therefore, we suggest the potential activation of the RAC1 pathway as a contributor to EAC tumorigenesis.
Figures
Similar articles
-
Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis.Nat Commun. 2014 Oct 29;5:5224. doi: 10.1038/ncomms6224. Nat Commun. 2014. PMID: 25351503 Free PMC article.
-
Novel Aberrations Uncovered in Barrett's Esophagus and Esophageal Adenocarcinoma Using Whole Transcriptome Sequencing.Mol Cancer Res. 2017 Nov;15(11):1558-1569. doi: 10.1158/1541-7786.MCR-17-0332. Epub 2017 Jul 27. Mol Cancer Res. 2017. PMID: 28751461
-
The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic.Nat Genet. 2019 Mar;51(3):506-516. doi: 10.1038/s41588-018-0331-5. Epub 2019 Feb 4. Nat Genet. 2019. PMID: 30718927 Free PMC article.
-
Genomic characterization of esophageal squamous cell carcinoma: Insights from next-generation sequencing.World J Gastroenterol. 2016 Feb 21;22(7):2284-93. doi: 10.3748/wjg.v22.i7.2284. World J Gastroenterol. 2016. PMID: 26900290 Free PMC article. Review.
-
MicroRNAs in Esophageal Adenocarcinoma: Functional Significance and Potential for the Development of New Molecular Disease Markers.Curr Pharm Des. 2015;21(23):3402-16. doi: 10.2174/1381612821666150311124418. Curr Pharm Des. 2015. PMID: 25758856 Review.
Cited by
-
Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy.Cancer Discov. 2015 Aug;5(8):821-831. doi: 10.1158/2159-8290.CD-15-0412. Epub 2015 May 23. Cancer Discov. 2015. PMID: 26003801 Free PMC article.
-
Understanding the cellular origin and progression of esophageal cancer using esophageal organoids.Cancer Lett. 2021 Jul 1;509:39-52. doi: 10.1016/j.canlet.2021.03.031. Epub 2021 Apr 7. Cancer Lett. 2021. PMID: 33838281 Free PMC article. Review.
-
Genomic similarity between gastroesophageal junction and esophageal Barrett's adenocarcinomas.Oncotarget. 2016 Aug 23;7(34):54867-54882. doi: 10.18632/oncotarget.10253. Oncotarget. 2016. PMID: 27363029 Free PMC article.
-
Docetaxel and its potential in the treatment of refractory esophagogastric adenocarcinoma.Therap Adv Gastroenterol. 2015 Jul;8(4):189-205. doi: 10.1177/1756283X15585468. Therap Adv Gastroenterol. 2015. PMID: 26136837 Free PMC article. Review.
-
Therapeutic advances of targeting receptor tyrosine kinases in cancer.Signal Transduct Target Ther. 2024 Aug 14;9(1):201. doi: 10.1038/s41392-024-01899-w. Signal Transduct Target Ther. 2024. PMID: 39138146 Free PMC article. Review.
References
-
- Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17:2–9. - PubMed
-
- Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142–6. - PubMed
-
- Wu AH, Wan P, Bernstein L. A multiethnic population-based study of smoking, alcohol and body size and risk of adenocarcinomas of the stomach and esophagus (United States) Cancer Causes Control. 2001;12:721–32. - PubMed
-
- Chung SM, Kao J, Hyjek E, Chen YT. p53 in esophageal adenocarcinoma: a critical reassessment of mutation frequency and identification of 72Arg as the dominant allele. Int J Oncol. 2007;31:1351–5. - PubMed
-
- Hardie LJ, et al. p16 expression in Barrett’s esophagus and esophageal adenocarcinoma: association with genetic and epigenetic alterations. Cancer Lett. 2005;217:221–30. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
