

Lost in transcription: molecular mechanisms that control HIV latency
- PMID: 23518577
- PMCID: PMC3705304
- DOI: 10.3390/v5030902
Lost in transcription: molecular mechanisms that control HIV latency
Abstract
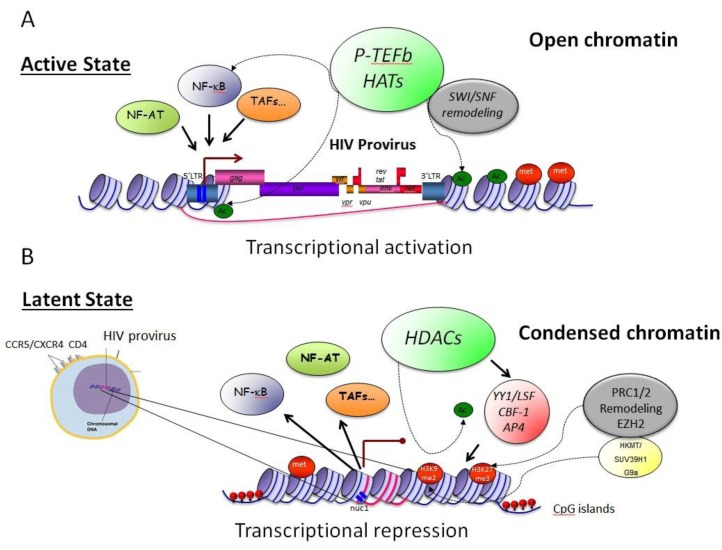
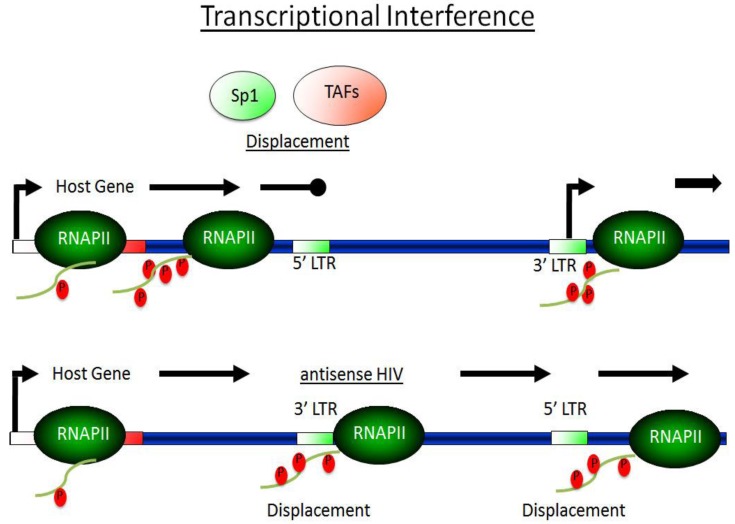
Highly active antiretroviral therapy (HAART) has limited the replication and spread of the human immunodeficiency virus (HIV). However, despite treatment, HIV infection persists in latently infected reservoirs, and once therapy is interrupted, viral replication rebounds quickly. Extensive efforts are being directed at eliminating these cell reservoirs. This feat can be achieved by reactivating latent HIV while administering drugs that prevent new rounds of infection and allow the immune system to clear the virus. However, current approaches to HIV eradication have not been effective. Moreover, as HIV latency is multifactorial, the significance of each of its molecular mechanisms is still under debate. Among these, transcriptional repression as a result of reduced levels and activity of the positive transcription elongation factor b (P-TEFb: CDK9/cyclin T) plays a significant role. Therefore, increasing levels of P-TEFb expression and activity is an excellent strategy to stimulate viral gene expression. This review summarizes the multiple steps that cause HIV to enter into latency. It positions the interplay between transcriptionally active and inactive host transcriptional activators and their viral partner Tat as valid targets for the development of new strategies to reactivate latent viral gene expression and eradicate HIV.
Figures


Similar articles
-
UHRF1 Suppresses HIV-1 Transcription and Promotes HIV-1 Latency by Competing with p-TEFb for Ubiquitination-Proteasomal Degradation of Tat.mBio. 2021 Aug 31;12(4):e0162521. doi: 10.1128/mBio.01625-21. Epub 2021 Aug 31. mBio. 2021. PMID: 34465029 Free PMC article.
-
Molecular mechanisms of HIV latency.J Clin Invest. 2016 Feb;126(2):448-54. doi: 10.1172/JCI80565. Epub 2016 Jan 5. J Clin Invest. 2016. PMID: 26731470 Free PMC article. Review.
-
Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3.mBio. 2018 Nov 13;9(6):e02158-18. doi: 10.1128/mBio.02158-18. mBio. 2018. PMID: 30425153 Free PMC article.
-
A chalcone derivative reactivates latent HIV-1 transcription through activating P-TEFb and promoting Tat-SEC interaction on viral promoter.Sci Rep. 2017 Sep 6;7(1):10657. doi: 10.1038/s41598-017-10728-w. Sci Rep. 2017. PMID: 28878233 Free PMC article.
-
The Molecular Basis for Human Immunodeficiency Virus Latency.Annu Rev Virol. 2017 Sep 29;4(1):261-285. doi: 10.1146/annurev-virology-101416-041646. Epub 2017 Jul 17. Annu Rev Virol. 2017. PMID: 28715973 Review.
Cited by
-
Impact of the Ku complex on HIV-1 expression and latency.PLoS One. 2013 Jul 29;8(7):e69691. doi: 10.1371/journal.pone.0069691. Print 2013. PLoS One. 2013. PMID: 23922776 Free PMC article.
-
Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies.Viruses. 2019 Mar 18;11(3):269. doi: 10.3390/v11030269. Viruses. 2019. PMID: 30889861 Free PMC article. Review.
-
Chemical probes targeting epigenetic proteins: Applications beyond oncology.Epigenetics. 2017 May 4;12(5):378-400. doi: 10.1080/15592294.2017.1279371. Epub 2017 Jan 12. Epigenetics. 2017. PMID: 28080202 Free PMC article. Review.
-
Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses.Vet Sci. 2015 Sep 29;2(4):293-348. doi: 10.3390/vetsci2040293. Vet Sci. 2015. PMID: 29061947 Free PMC article. Review.
-
Heterogeneity of Latency Establishment in the Different Human CD4+ T Cell Subsets Stimulated with IL-15.J Virol. 2022 May 25;96(10):e0037922. doi: 10.1128/jvi.00379-22. Epub 2022 May 2. J Virol. 2022. PMID: 35499323 Free PMC article.
References
-
- Chun T.W., Carruth L., Finzi D., Shen X., DiGiuseppe J.A., Taylor H., Hermankova M., Chadwick K., Margolick J., Quinn T.C., et al. Quantification of latent tissue reservoirs and total body viral load in HIV–1 infection. Nature. 1997;387:183–188. - PubMed
-
- Prins J.M., Jurriaans S., van Praag R.M., Blaak H., van Rij R., Schellekens P.T., ten Berge I.J., Yong S.L., Fox C.H., Roos M.T., et al. Immuno–activation with anti–CD3 and recombinant human IL–2 in HIV–1–infected patients on potent antiretroviral therapy. AIDS. 1999;13:2405–2410. doi: 10.1097/00002030-199912030-00012. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
