

Barth syndrome
- PMID: 23398819
- PMCID: PMC3583704
- DOI: 10.1186/1750-1172-8-23
Barth syndrome
Abstract
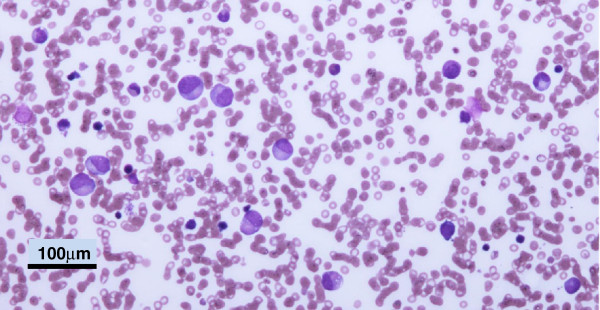
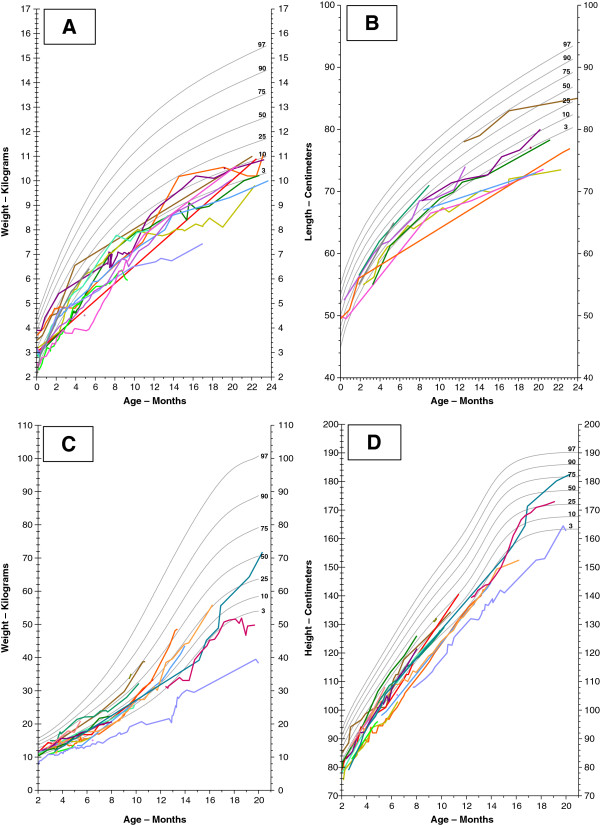
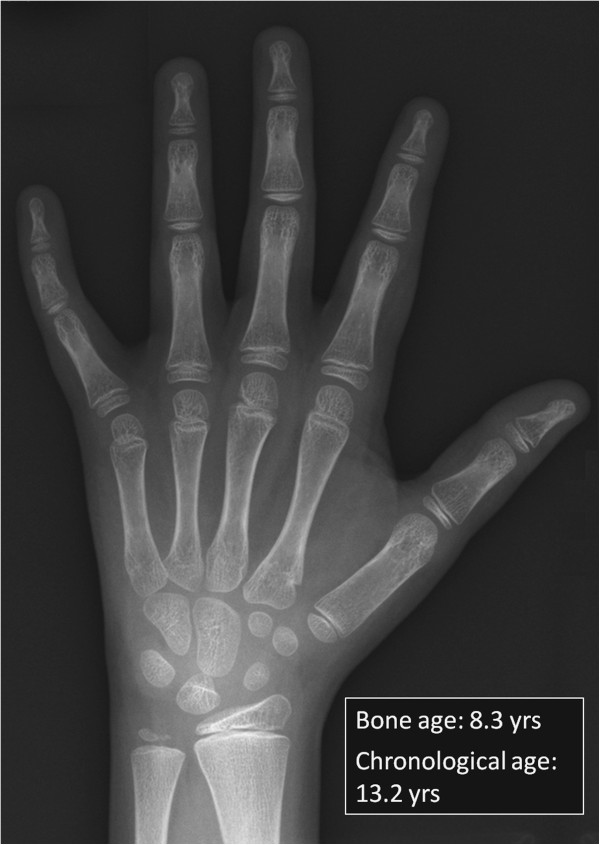
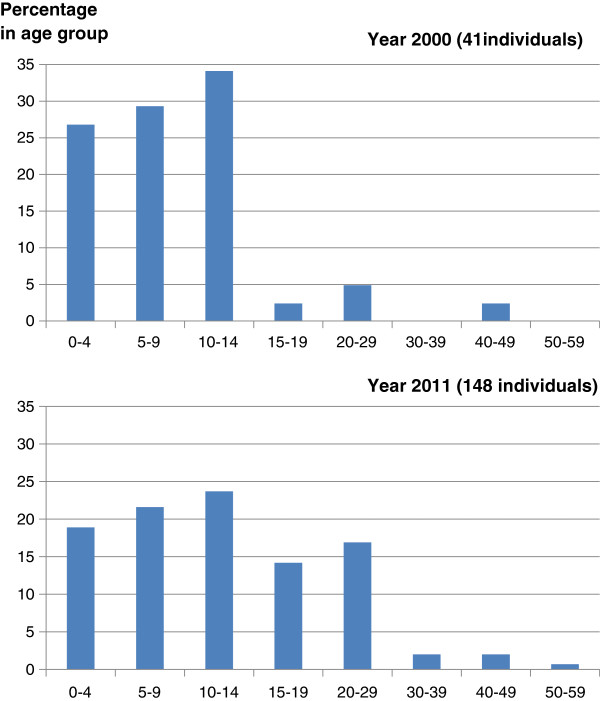
First described in 1983, Barth syndrome (BTHS) is widely regarded as a rare X-linked genetic disease characterised by cardiomyopathy (CM), skeletal myopathy, growth delay, neutropenia and increased urinary excretion of 3-methylglutaconic acid (3-MGCA). Fewer than 200 living males are known worldwide, but evidence is accumulating that the disorder is substantially under-diagnosed. Clinical features include variable combinations of the following wide spectrum: dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), endocardial fibroelastosis (EFE), left ventricular non-compaction (LVNC), ventricular arrhythmia, sudden cardiac death, prolonged QTc interval, delayed motor milestones, proximal myopathy, lethargy and fatigue, neutropenia (absent to severe; persistent, intermittent or perfectly cyclical), compensatory monocytosis, recurrent bacterial infection, hypoglycaemia, lactic acidosis, growth and pubertal delay, feeding problems, failure to thrive, episodic diarrhoea, characteristic facies, and X-linked family history. Historically regarded as a cardiac disease, BTHS is now considered a multi-system disorder which may be first seen by many different specialists or generalists. Phenotypic breadth and variability present a major challenge to the diagnostician: some children with BTHS have never been neutropenic, whereas others lack increased 3-MGCA and a minority has occult or absent CM. Furthermore, BTHS was first described in 2010 as an unrecognised cause of fetal death. Disabling mutations or deletions of the tafazzin (TAZ) gene, located at Xq28, cause the disorder by reducing remodeling of cardiolipin, a principal phospholipid of the inner mitochondrial membrane. A definitive biochemical test, based on detecting abnormal ratios of different cardiolipin species, was first described in 2008. Key areas of differential diagnosis include metabolic and viral cardiomyopathies, mitochondrial diseases, and many causes of neutropenia and recurrent male miscarriage and stillbirth. Cardiolipin testing and TAZ sequencing now provide relatively rapid diagnostic testing, both prospectively and retrospectively, from a range of fresh or stored tissues, blood or neonatal bloodspots. TAZ sequencing also allows female carrier detection and antenatal screening. Management of BTHS includes medical therapy of CM, cardiac transplantation (in 14% of patients), antibiotic prophylaxis and granulocyte colony-stimulating factor (G-CSF) therapy. Multidisciplinary teams/clinics are essential for minimising hospital attendances and allowing many more individuals with BTHS to live into adulthood.
Figures

Similar articles
-
Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle.Mol Genet Metab. 2012 Nov;107(3):428-32. doi: 10.1016/j.ymgme.2012.09.013. Epub 2012 Sep 18. Mol Genet Metab. 2012. PMID: 23031367 Free PMC article.
-
Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth.Prenat Diagn. 2010 Oct;30(10):970-6. doi: 10.1002/pd.2599. Prenat Diagn. 2010. PMID: 20812380 Free PMC article.
-
Barth syndrome cardiomyopathy: targeting the mitochondria with elamipretide.Heart Fail Rev. 2021 Mar;26(2):237-253. doi: 10.1007/s10741-020-10031-3. Epub 2020 Oct 1. Heart Fail Rev. 2021. PMID: 33001359 Free PMC article. Review.
-
Left ventricular noncompaction (LVNC) and low mitochondrial membrane potential are specific for Barth syndrome.J Inherit Metab Dis. 2013 Nov;36(6):929-37. doi: 10.1007/s10545-013-9584-4. Epub 2013 Jan 30. J Inherit Metab Dis. 2013. PMID: 23361305 Free PMC article.
-
Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy.Lipids. 2017 Feb;52(2):99-108. doi: 10.1007/s11745-016-4229-7. Epub 2017 Jan 9. Lipids. 2017. PMID: 28070695 Free PMC article. Review.
Cited by
-
Increased Reactive Oxygen Species-Mediated Ca2+/Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome.Circulation. 2021 May 11;143(19):1894-1911. doi: 10.1161/CIRCULATIONAHA.120.048698. Epub 2021 Apr 1. Circulation. 2021. PMID: 33793303 Free PMC article.
-
Cancer as a mitochondrial metabolic disease.Front Cell Dev Biol. 2015 Jul 7;3:43. doi: 10.3389/fcell.2015.00043. eCollection 2015. Front Cell Dev Biol. 2015. PMID: 26217661 Free PMC article.
-
A novel TAZ gene mutation and mosaicism in a Polish family with Barth syndrome.Ann Hum Genet. 2015 May;79(3):218-24. doi: 10.1111/ahg.12108. Epub 2015 Mar 16. Ann Hum Genet. 2015. PMID: 25776009 Free PMC article.
-
Mitochondrial Dysfunctions: Genetic and Cellular Implications Revealed by Various Model Organisms.Genes (Basel). 2024 Sep 1;15(9):1153. doi: 10.3390/genes15091153. Genes (Basel). 2024. PMID: 39336744 Free PMC article. Review.
-
The Impact of Raising Children with Barth Syndrome on Parental Health-Related Quality of Life and Family Functioning: Preliminary Reliability and Validity of the PedsQL™ Family Impact Module.Occup Ther Int. 2023 Dec 30;2023:5588935. doi: 10.1155/2023/5588935. eCollection 2023. Occup Ther Int. 2023. PMID: 38187035 Free PMC article.
References
-
- Barth PG, Scholte HR, Berden JA, Vanderkleivanmoorsel JM, Luythouwen IEM, Vantveerkorthof ET, Vanderharten JJ, Sobotkaplojhar MA. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62:327–355. doi: 10.1016/0022-510X(83)90209-5. - DOI - PubMed
-
- Neustein HB, Lurie PR, Dahms B, Takahashi M. An X-linked recessive cardiomyopathy with abnormal mitochondria. Pediatrics. 1979;64:24–29. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
