

Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells
- PMID: 23363601
- PMCID: PMC3596255
- DOI: 10.1091/mbc.E12-10-0747
Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells
Abstract
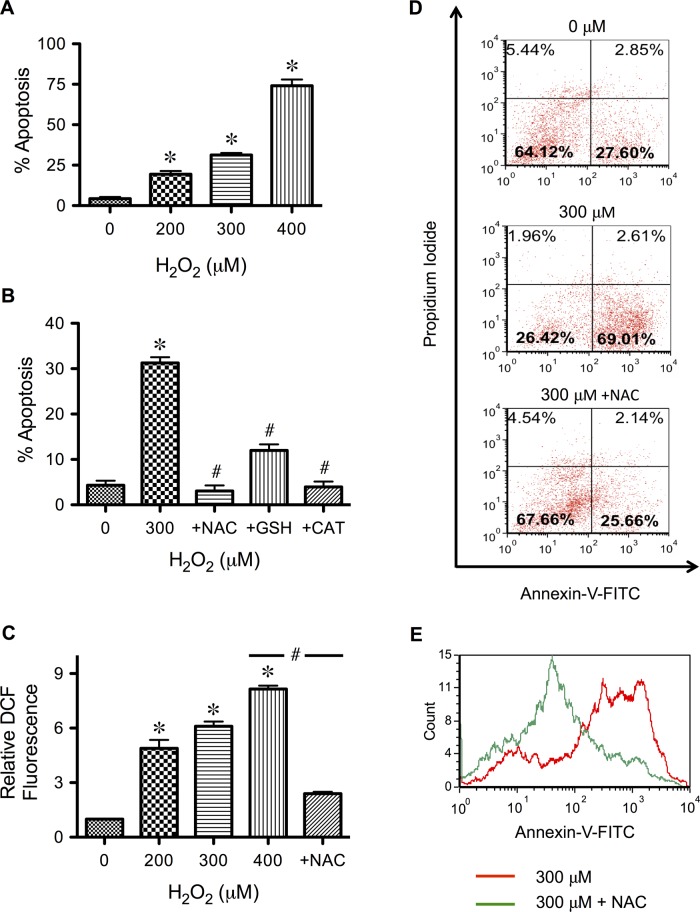
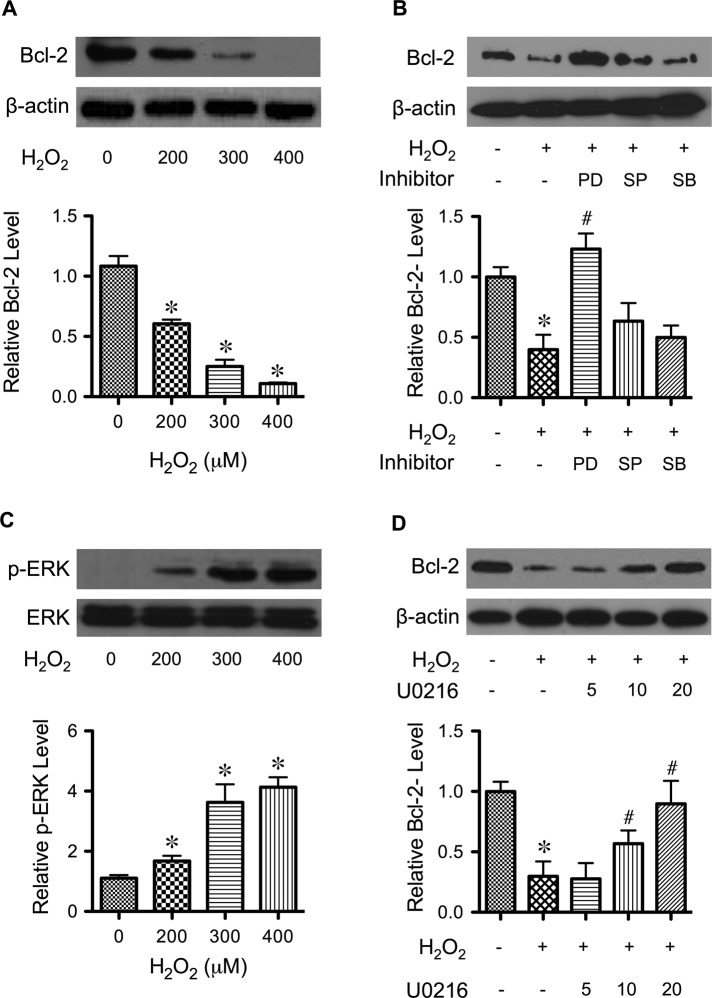
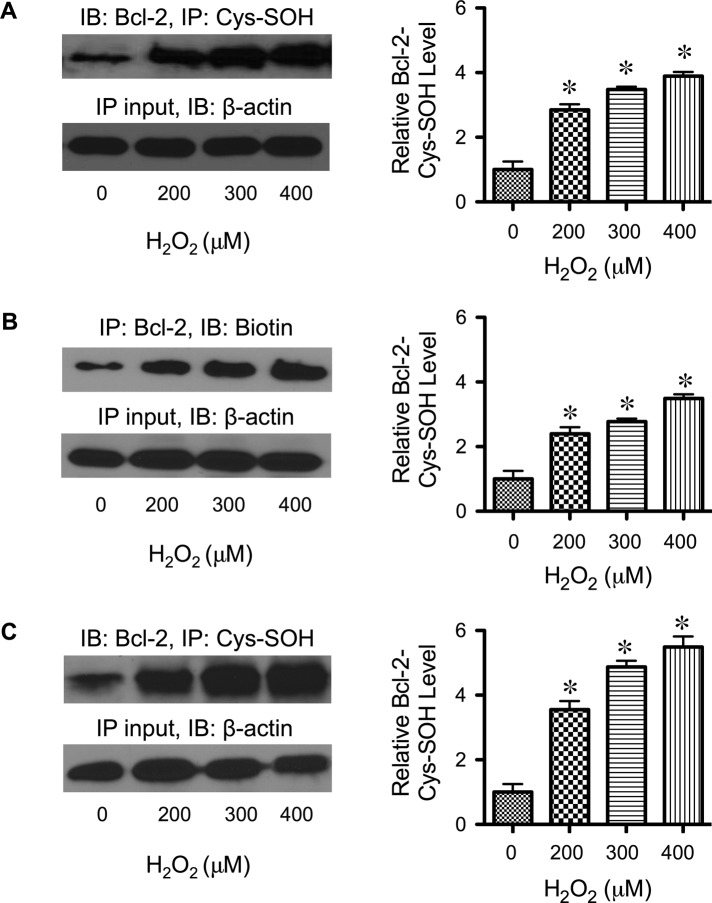
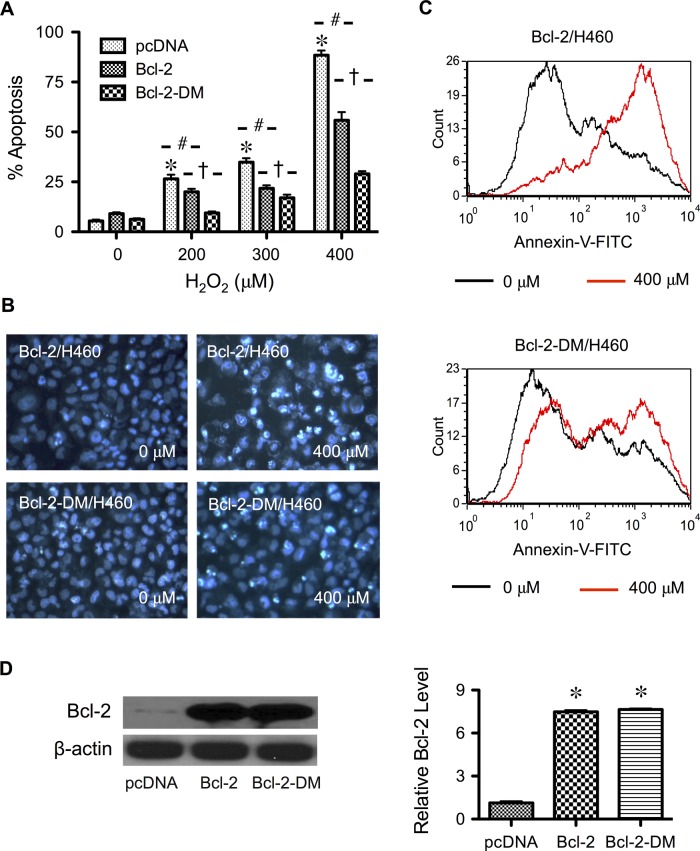
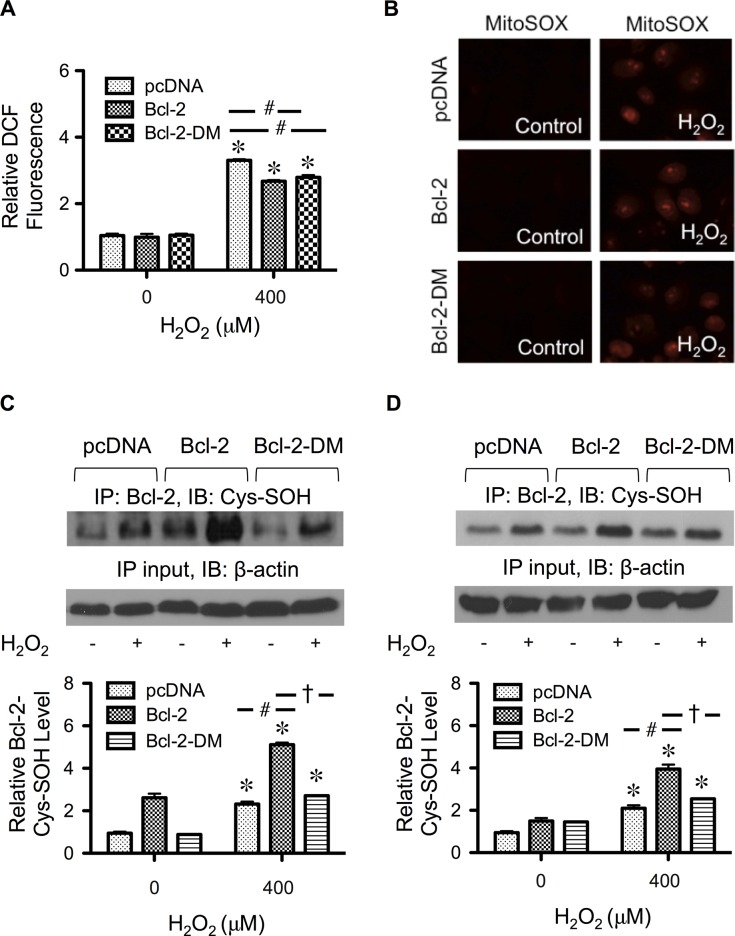
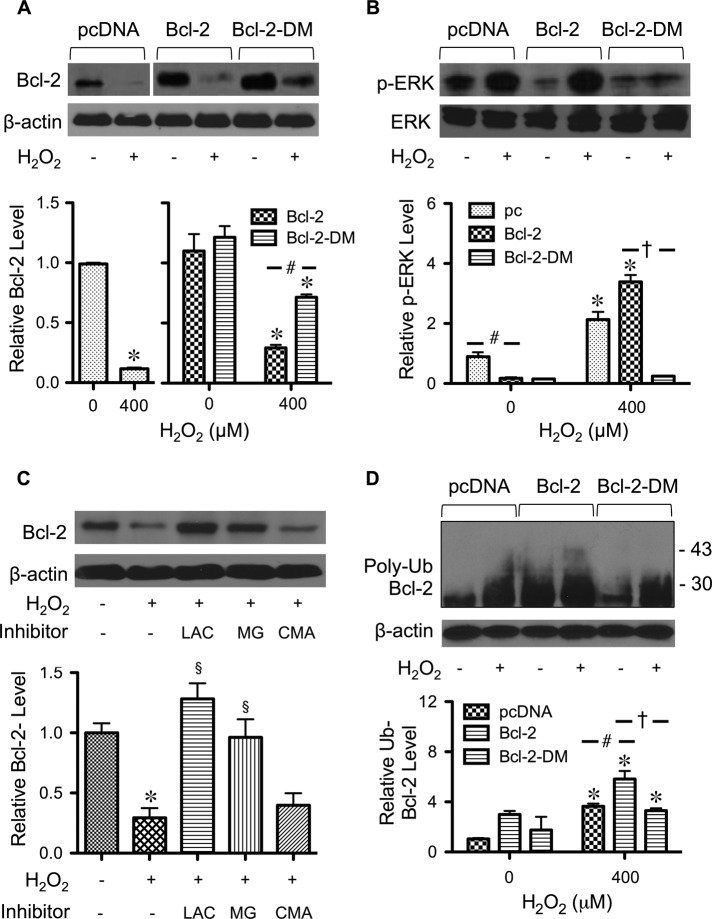
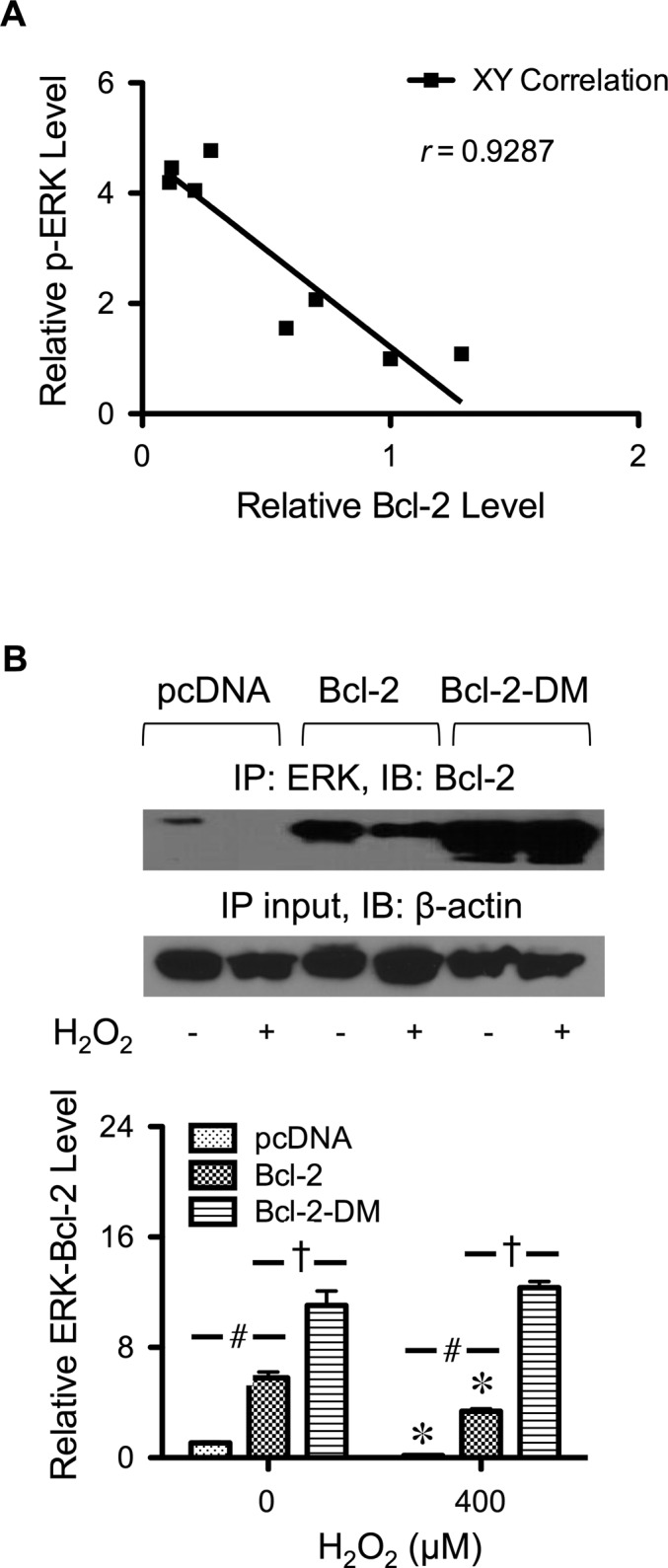
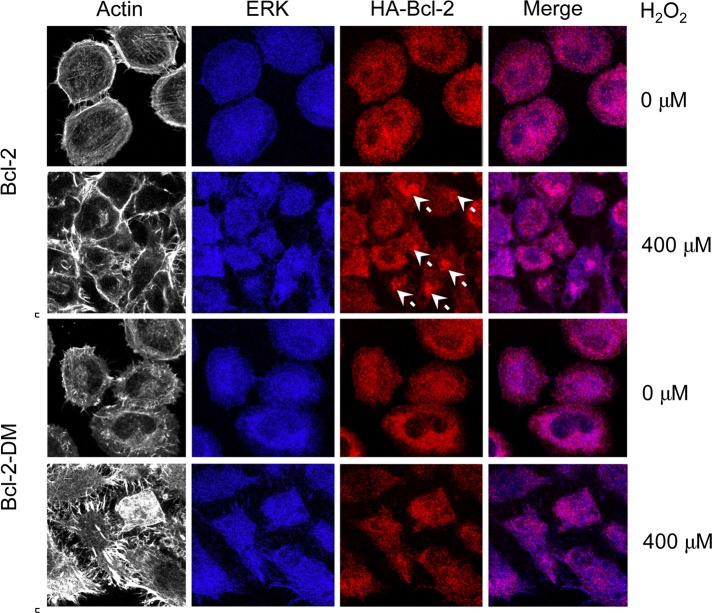
Hydrogen peroxide is a key mediator of oxidative stress known to be important in various cellular processes, including apoptosis. B-cell lymphoma-2 (Bcl-2) is an oxidative stress-responsive protein and a key regulator of apoptosis; however, the underlying mechanisms of oxidative regulation of Bcl-2 are not well understood. The present study investigates the direct effect of H2O2 on Bcl-2 cysteine oxidation as a potential mechanism of apoptosis regulation. Exposure of human lung epithelial cells to H2O2 induces apoptosis concomitant with cysteine oxidation and down-regulation of Bcl-2. Inhibition of Bcl-2 oxidation by antioxidants or by site-directed mutagenesis of Bcl-2 at Cys-158 and Cys-229 abrogates the effects of H2O2 on Bcl-2 and apoptosis. Immunoprecipitation and confocal microscopic studies show that Bcl-2 interacts with mitogen-activated protein kinase (extracellular signal-regulated kinase 1/2 [ERK1/2]) to suppress apoptosis and that this interaction is modulated by cysteine oxidation of Bcl-2. The H2O2-induced Bcl-2 cysteine oxidation interferes with Bcl-2 and ERK1/2 interaction. Mutation of the cysteine residues inhibits the disruption of Bcl-2-ERK complex, as well as the induction of apoptosis by H2O2. Taken together, these results demonstrate the critical role of Bcl-2 cysteine oxidation in the regulation of apoptosis through ERK signaling. This new finding reveals crucial redox regulatory mechanisms that control the antiapoptotic function of Bcl-2.
Figures
Similar articles
-
H2O2 oxidation of cysteine residues in c-Jun N-terminal kinase 2 (JNK2) contributes to redox regulation in human articular chondrocytes.J Biol Chem. 2018 Oct 19;293(42):16376-16389. doi: 10.1074/jbc.RA118.004613. Epub 2018 Sep 6. J Biol Chem. 2018. PMID: 30190325 Free PMC article.
-
Norepinephrine induces apoptosis in neonatal rat endothelial cells via a ROS-dependent JNK activation pathway.Apoptosis. 2006 Nov;11(11):2053-63. doi: 10.1007/s10495-006-0192-8. Apoptosis. 2006. PMID: 17041759
-
Oxidative stress attenuates Fas-mediated apoptosis in Jurkat T cell line through Bfl-1 induction.Oncogene. 2005 Feb 10;24(7):1252-61. doi: 10.1038/sj.onc.1208282. Oncogene. 2005. PMID: 15592513
-
Peroxiporins and Oxidative Stress: Promising Targets to Tackle Inflammation and Cancer.Int J Mol Sci. 2024 Aug 1;25(15):8381. doi: 10.3390/ijms25158381. Int J Mol Sci. 2024. PMID: 39125952 Free PMC article. Review.
-
Redox regulation of protein kinases.Crit Rev Biochem Mol Biol. 2013 Jul-Aug;48(4):332-56. doi: 10.3109/10409238.2013.790873. Epub 2013 May 3. Crit Rev Biochem Mol Biol. 2013. PMID: 23639002 Free PMC article. Review.
Cited by
-
The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival.Cells. 2021 Sep 13;10(9):2401. doi: 10.3390/cells10092401. Cells. 2021. PMID: 34572050 Free PMC article. Review.
-
TIGAR contributes to ischemic tolerance induced by cerebral preconditioning through scavenging of reactive oxygen species and inhibition of apoptosis.Sci Rep. 2016 Jun 3;6:27096. doi: 10.1038/srep27096. Sci Rep. 2016. PMID: 27256465 Free PMC article.
-
Phlorotannins: Novel Orally Administrated Bioactive Compounds That Induce Mitochondrial Dysfunction and Oxidative Stress in Cancer.Antioxidants (Basel). 2023 Sep 7;12(9):1734. doi: 10.3390/antiox12091734. Antioxidants (Basel). 2023. PMID: 37760037 Free PMC article. Review.
-
SLUG is required for SOX9 stabilization and functions to promote cancer stem cells and metastasis in human lung carcinoma.Oncogene. 2016 Jun 2;35(22):2824-33. doi: 10.1038/onc.2015.351. Epub 2015 Sep 21. Oncogene. 2016. PMID: 26387547 Free PMC article.
-
Oxidative Stress-Mediated Programmed Cell Death: a Potential Therapy Target for Atherosclerosis.Cardiovasc Drugs Ther. 2024 Aug;38(4):819-832. doi: 10.1007/s10557-022-07414-z. Epub 2022 Dec 16. Cardiovasc Drugs Ther. 2024. PMID: 36522550 Review.
References
-
- Chanvorachote P, Nimmanit U, Stehlik C, Wang L, Jiang B, Ongpipatanakul B, Rojanasakul Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006;66:6353–6360. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
