

Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice
- PMID: 23281774
- PMCID: PMC3538568
- DOI: 10.1186/1750-1326-8-1
Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice
Abstract
Background: A proline-to-serine substitution at position-56 (P56S) of vesicle-associated membrane protein-associated protein B (VAPB) causes a form of dominantly inherited motor neuron disease (MND), including typical and atypical amyotrophic lateral sclerosis (ALS) and a mild late-onset spinal muscular atrophy (SMA). VAPB is an integral endoplasmic reticulum (ER) protein and has been implicated in various cellular processes, including ER stress, the unfolded protein response (UPR) and Ca2+ homeostasis. However, it is unclear how the P56S mutation leads to neurodegeneration and muscle atrophy in patients. The formation of abnormal VAPB-positive inclusions by mutant VAPB suggests a possible toxic gain of function as an underlying mechanism. Furthermore, the amount of VAPB protein is reported to be reduced in sporadic ALS patients and mutant SOD1G93A mice, leading to the hypothesis that wild type VAPB plays a role in the pathogenesis of ALS without VAPB mutations.
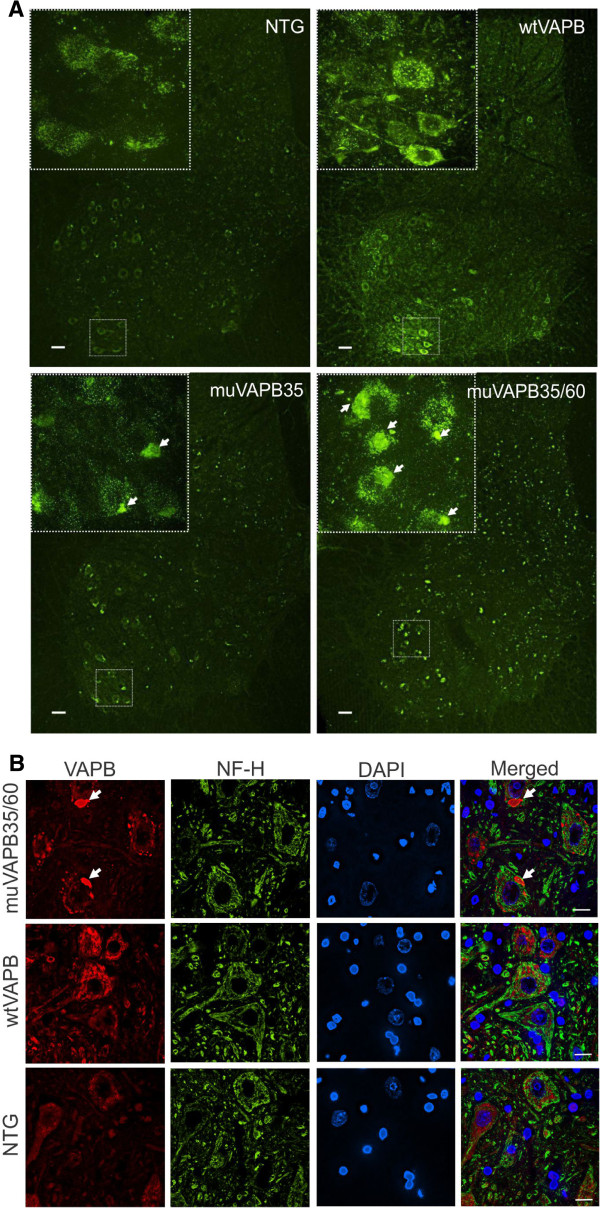
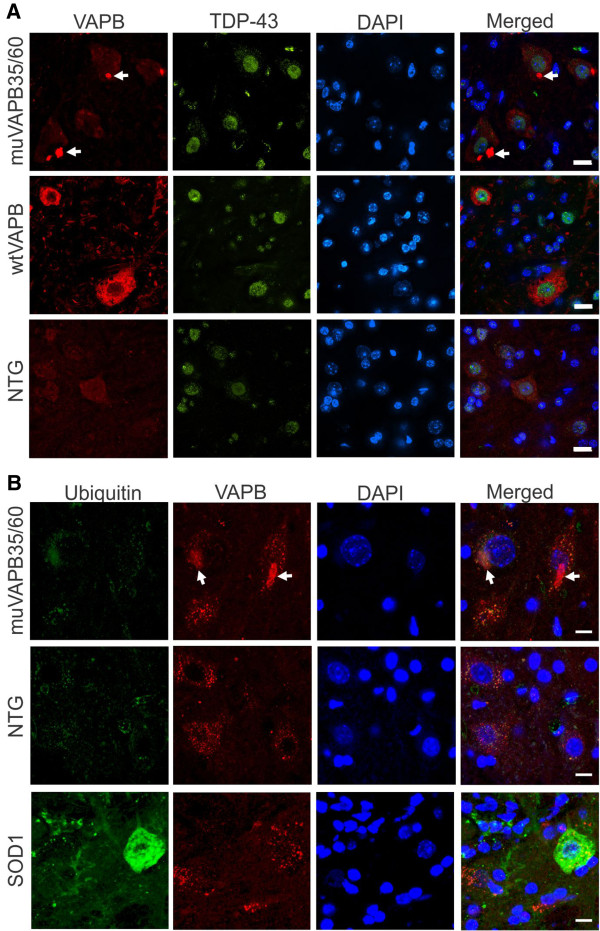
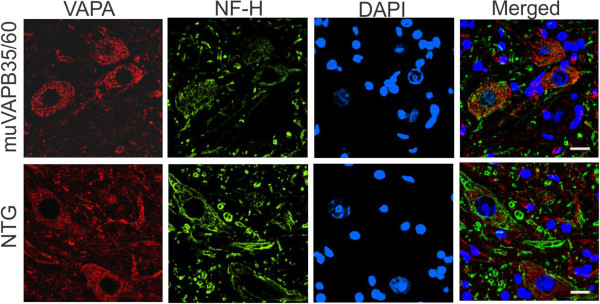
Results: To investigate the pathogenic mechanism in vivo, we generated human wild type (wtVAPB) and mutant VAPB (muVAPB) transgenic mice that expressed the transgenes broadly in the CNS. We observed robust VAPB-positive aggregates in the spinal cord of muVAPB transgenic mice. However, we failed to find an impairment of motor function and motor neuron degeneration. We also did not detect any change in the endogenous VAPB level or evidence for induction of the unfolded protein response (UPR) and coaggregation of VAPA with muVAPB. Furthermore, we crossed these VAPB transgenic mice with mice that express mutant SOD1G93A and develop motor neuron degeneration. Overexpression of neither wtVAPB nor muVAPB modulated the protein aggregation and disease progression in the SOD1G93A mice.
Conclusion: Overexpression of VAPBP56S mutant to approximately two-fold of the endogenous VAPB in mouse spinal cord produced abundant VAPB aggregates but was not sufficient to cause motor dysfunction or motor neuron degeneration. Furthermore, overexpression of either muVAPB or wtVAPB does not modulate the course of ALS in SOD1G93A mice. These results suggest that changes in wild type VAPB do not play a significant role in ALS cases that are not caused by VAPB mutations. Furthermore, these results suggest that muVAPB aggregates are innocuous and do not cause motor neuron degeneration by a gain-of-toxicity, and therefore, a loss of function may be the underlying mechanism.
Figures







Similar articles
-
Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates.J Neurosci. 2007 Sep 5;27(36):9801-15. doi: 10.1523/JNEUROSCI.2661-07.2007. J Neurosci. 2007. PMID: 17804640 Free PMC article.
-
Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons.Hum Mol Genet. 2013 Nov 1;22(21):4293-305. doi: 10.1093/hmg/ddt279. Epub 2013 Jun 13. Hum Mol Genet. 2013. PMID: 23771029 Free PMC article.
-
Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response.Hum Mol Genet. 2015 Nov 15;24(22):6515-29. doi: 10.1093/hmg/ddv360. Epub 2015 Sep 11. Hum Mol Genet. 2015. PMID: 26362257 Free PMC article.
-
Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis.IUBMB Life. 2011 Sep;63(9):754-63. doi: 10.1002/iub.520. Epub 2011 Aug 10. IUBMB Life. 2011. PMID: 21834058 Review.
-
ER stress and unfolded protein response in amyotrophic lateral sclerosis.Mol Neurobiol. 2009 Apr;39(2):81-9. doi: 10.1007/s12035-009-8054-3. Epub 2009 Jan 30. Mol Neurobiol. 2009. PMID: 19184563 Review.
Cited by
-
Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan.Hum Mol Genet. 2015 Aug 1;24(15):4353-64. doi: 10.1093/hmg/ddv168. Epub 2015 May 10. Hum Mol Genet. 2015. PMID: 25964428 Free PMC article.
-
VAP Proteins - From Organelle Tethers to Pathogenic Host Interactors and Their Role in Neuronal Disease.Front Cell Dev Biol. 2022 Jun 8;10:895856. doi: 10.3389/fcell.2022.895856. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35756994 Free PMC article. Review.
-
Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment.Acta Neuropathol Commun. 2013 Jun 12;1:24. doi: 10.1186/2051-5960-1-24. Acta Neuropathol Commun. 2013. PMID: 24252306 Free PMC article.
-
Potential roles of the endoplasmic reticulum stress pathway in amyotrophic lateral sclerosis.Front Aging Neurosci. 2023 Feb 15;15:1047897. doi: 10.3389/fnagi.2023.1047897. eCollection 2023. Front Aging Neurosci. 2023. PMID: 36875699 Free PMC article. Review.
-
SOD1 activity threshold and TOR signalling modulate VAP(P58S) aggregation via reactive oxygen species-induced proteasomal degradation in a Drosophila model of amyotrophic lateral sclerosis.Dis Model Mech. 2019 Feb 7;12(2):dmm033803. doi: 10.1242/dmm.033803. Dis Model Mech. 2019. PMID: 30635270 Free PMC article.
References
-
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J. et al.A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
