

Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions
- PMID: 23270813
- PMCID: PMC3644557
- DOI: 10.1016/j.ymeth.2012.12.005
Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions
Abstract
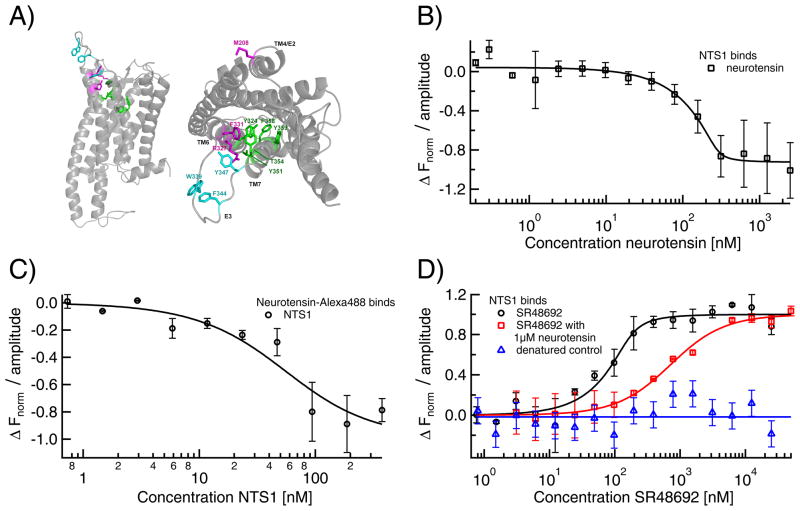
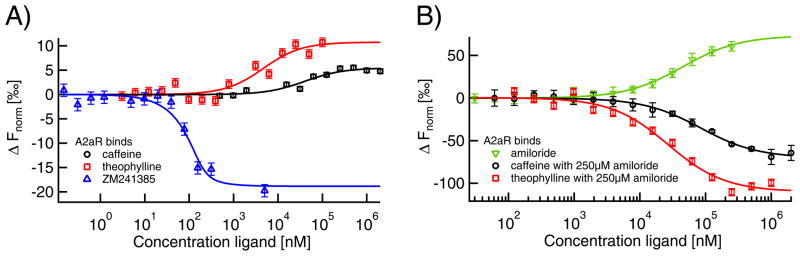
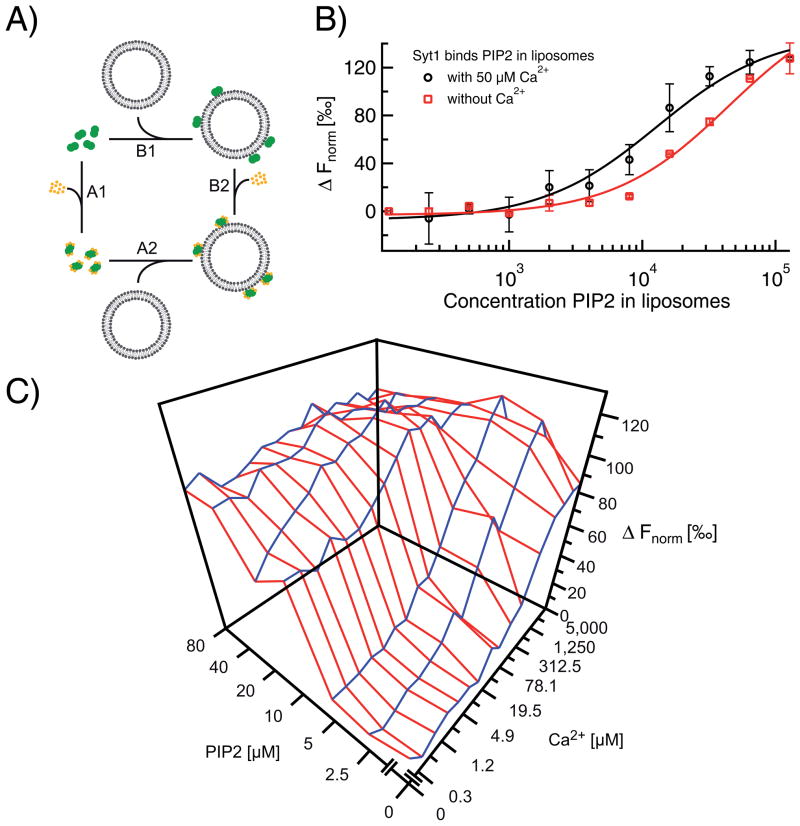
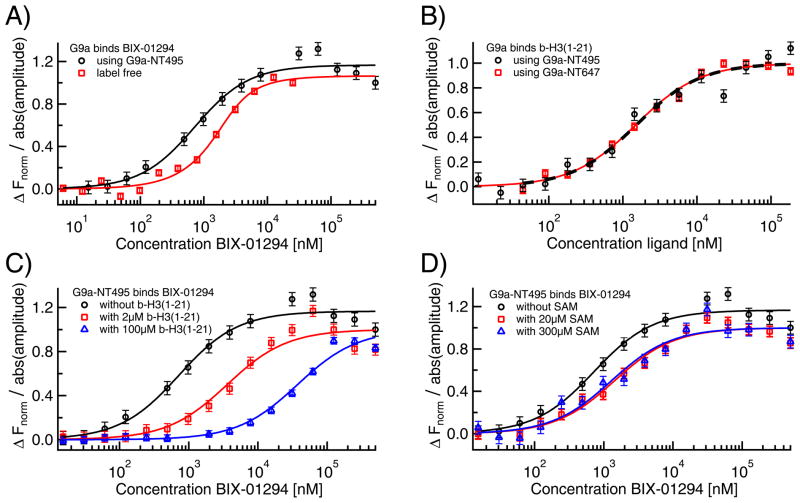
Microscale thermophoresis (MST) allows for quantitative analysis of protein interactions in free solution and with low sample consumption. The technique is based on thermophoresis, the directed motion of molecules in temperature gradients. Thermophoresis is highly sensitive to all types of binding-induced changes of molecular properties, be it in size, charge, hydration shell or conformation. In an all-optical approach, an infrared laser is used for local heating, and molecule mobility in the temperature gradient is analyzed via fluorescence. In standard MST one binding partner is fluorescently labeled. However, MST can also be performed label-free by exploiting intrinsic protein UV-fluorescence. Despite the high molecular weight ratio, the interaction of small molecules and peptides with proteins is readily accessible by MST. Furthermore, MST assays are highly adaptable to fit to the diverse requirements of different biomolecules, such as membrane proteins to be stabilized in solution. The type of buffer and additives can be chosen freely. Measuring is even possible in complex bioliquids like cell lysate allowing close to in vivo conditions without sample purification. Binding modes that are quantifiable via MST include dimerization, cooperativity and competition. Thus, its flexibility in assay design qualifies MST for analysis of biomolecular interactions in complex experimental settings, which we herein demonstrate by addressing typically challenging types of binding events from various fields of life science.
Copyright © 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest statement: Stefan Duhr and Philipp Baaske are founders, Moran Jerabek-Willemsen is an employee of the LMU spin-off company NanoTemper Technologies GmbH, which provides services and devices based on MST.
Figures




Similar articles
-
Studying small molecule-aptamer interactions using MicroScale Thermophoresis (MST).Methods. 2016 Mar 15;97:27-34. doi: 10.1016/j.ymeth.2015.08.023. Epub 2015 Aug 31. Methods. 2016. PMID: 26334574
-
Mapping the Binding Site of an Aptamer on ATP Using MicroScale Thermophoresis.J Vis Exp. 2017 Jan 7;(119):55070. doi: 10.3791/55070. J Vis Exp. 2017. PMID: 28117825 Free PMC article.
-
Measurement of Protein-Protein Interactions through Microscale Thermophoresis (MST).Bio Protoc. 2020 Apr 5;10(7):e3574. doi: 10.21769/BioProtoc.3574. eCollection 2020 Apr 5. Bio Protoc. 2020. PMID: 33659544 Free PMC article.
-
Molecular interaction studies using microscale thermophoresis.Assay Drug Dev Technol. 2011 Aug;9(4):342-53. doi: 10.1089/adt.2011.0380. Assay Drug Dev Technol. 2011. PMID: 21812660 Free PMC article. Review.
-
Thermophoresis for characterizing biomolecular interaction.Methods. 2018 Aug 15;146:107-119. doi: 10.1016/j.ymeth.2018.02.003. Epub 2018 Feb 10. Methods. 2018. PMID: 29438829 Review.
Cited by
-
Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility.Elife. 2014 Dec 23;3:e04008. doi: 10.7554/eLife.04008. Elife. 2014. PMID: 25534642 Free PMC article.
-
RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma.Sci Adv. 2020 Jul 17;6(29):eaba2113. doi: 10.1126/sciadv.aba2113. eCollection 2020 Jul. Sci Adv. 2020. PMID: 32832624 Free PMC article.
-
The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination.Theranostics. 2020 Aug 8;10(22):10141-10153. doi: 10.7150/thno.46985. eCollection 2020. Theranostics. 2020. PMID: 32929339 Free PMC article. Review.
-
Microscale thermophoresis as a powerful tool for screening glycosyltransferases involved in cell wall biosynthesis.Plant Methods. 2020 Jul 28;16:99. doi: 10.1186/s13007-020-00641-1. eCollection 2020. Plant Methods. 2020. PMID: 32742297 Free PMC article.
-
Approved Anti-cancer Drugs Target Oncogenic Non-coding RNAs.Cell Chem Biol. 2018 Sep 20;25(9):1086-1094.e7. doi: 10.1016/j.chembiol.2018.05.015. Epub 2018 Jun 28. Cell Chem Biol. 2018. PMID: 30251629 Free PMC article.
References
-
- Lin CC, Melo FA, Ghosh R, Suen KM, Stagg LJ, Kirkpatrick J, Arold ST, Ahmed Z, Ladbury JE. Inhibition of Basal FGF Receptor Signaling by Dimeric Grb2. Cell. 2012;149:1514–1524. - PubMed
-
- Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
