

Intracellular vesicle acidification promotes maturation of infectious poliovirus particles
- PMID: 23209416
- PMCID: PMC3510256
- DOI: 10.1371/journal.ppat.1003046
Intracellular vesicle acidification promotes maturation of infectious poliovirus particles
Abstract
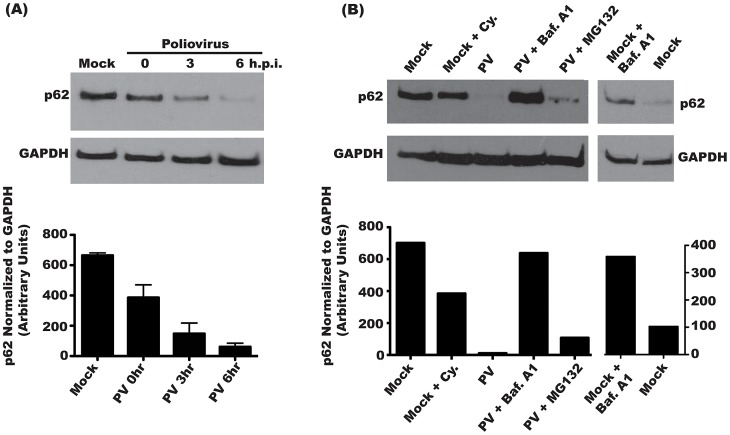
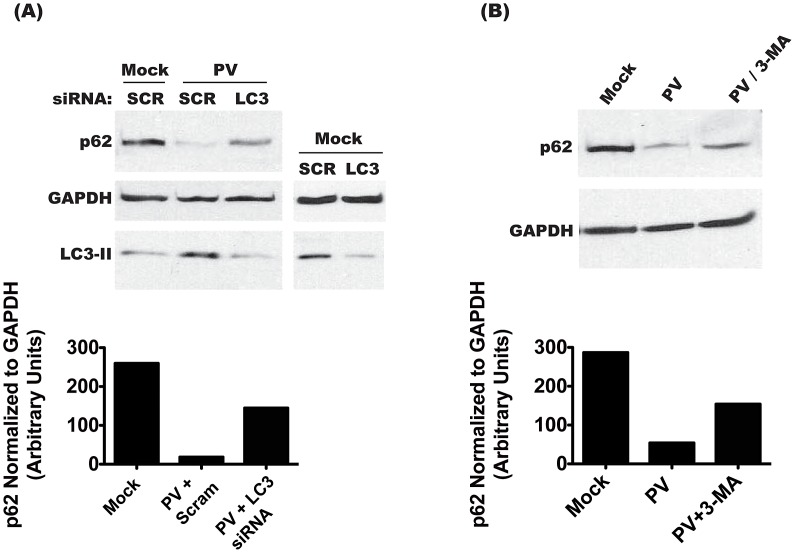
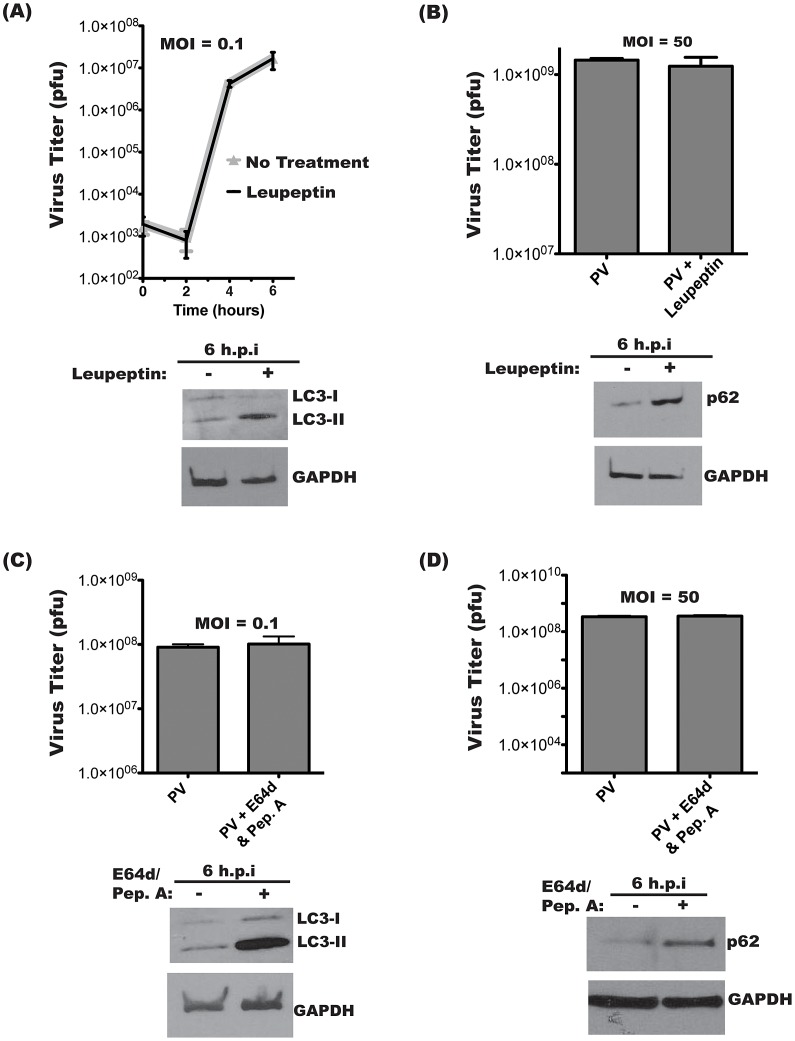
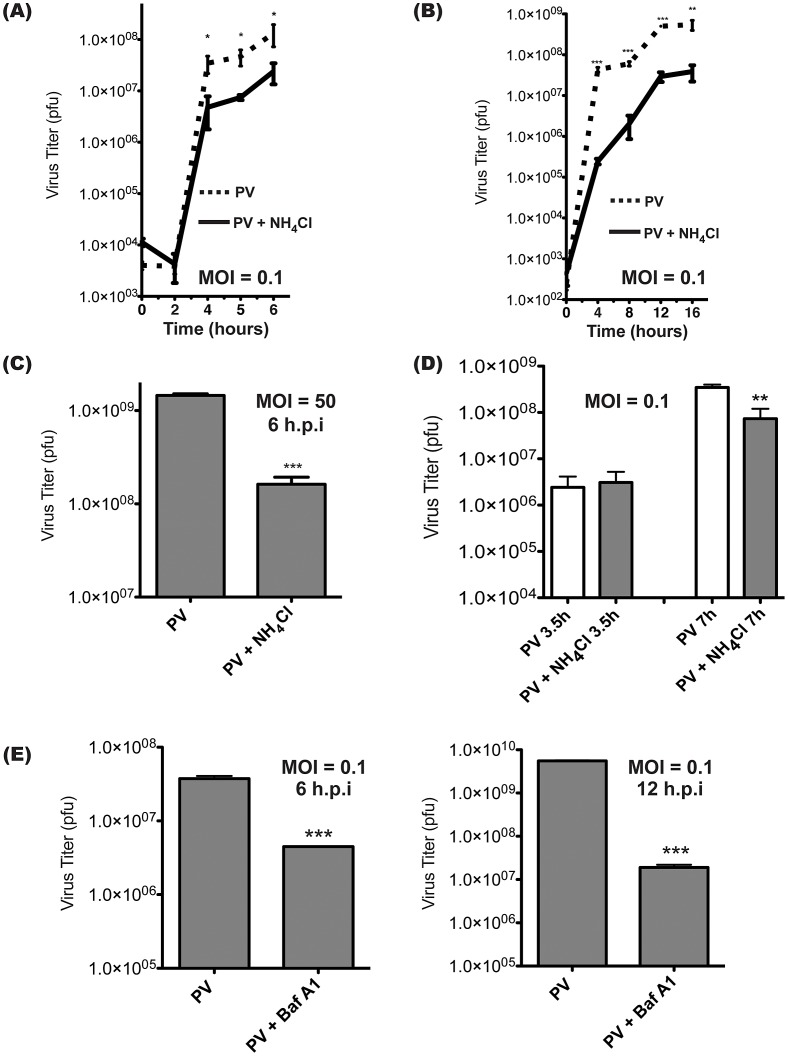
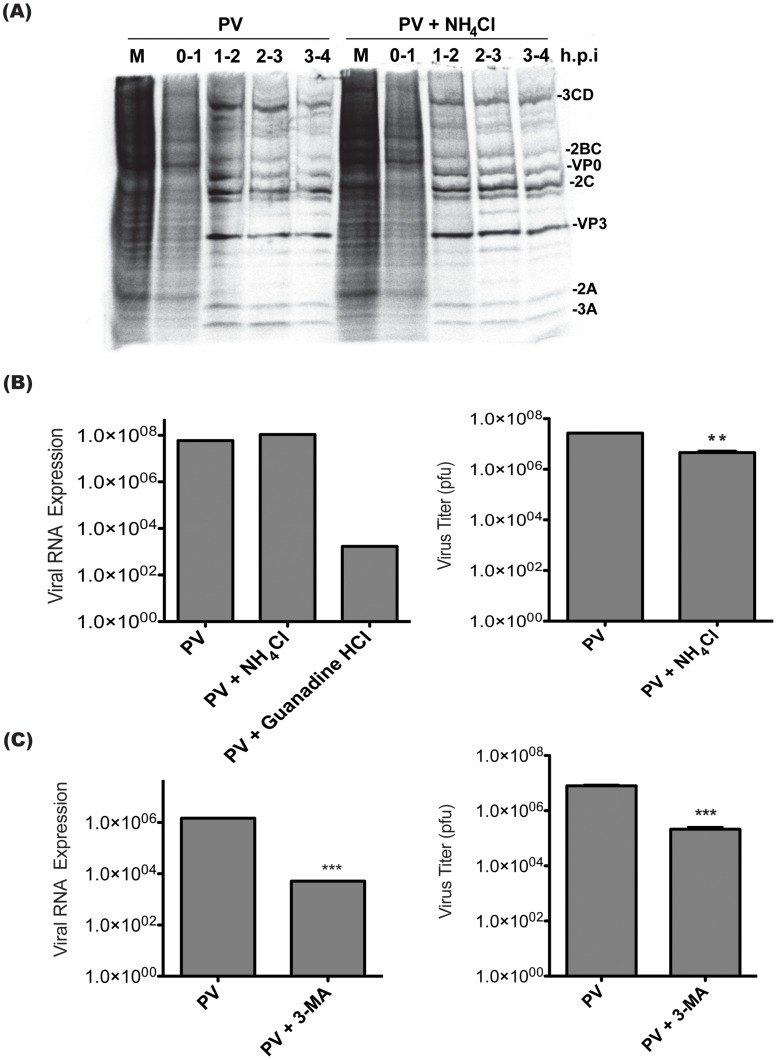
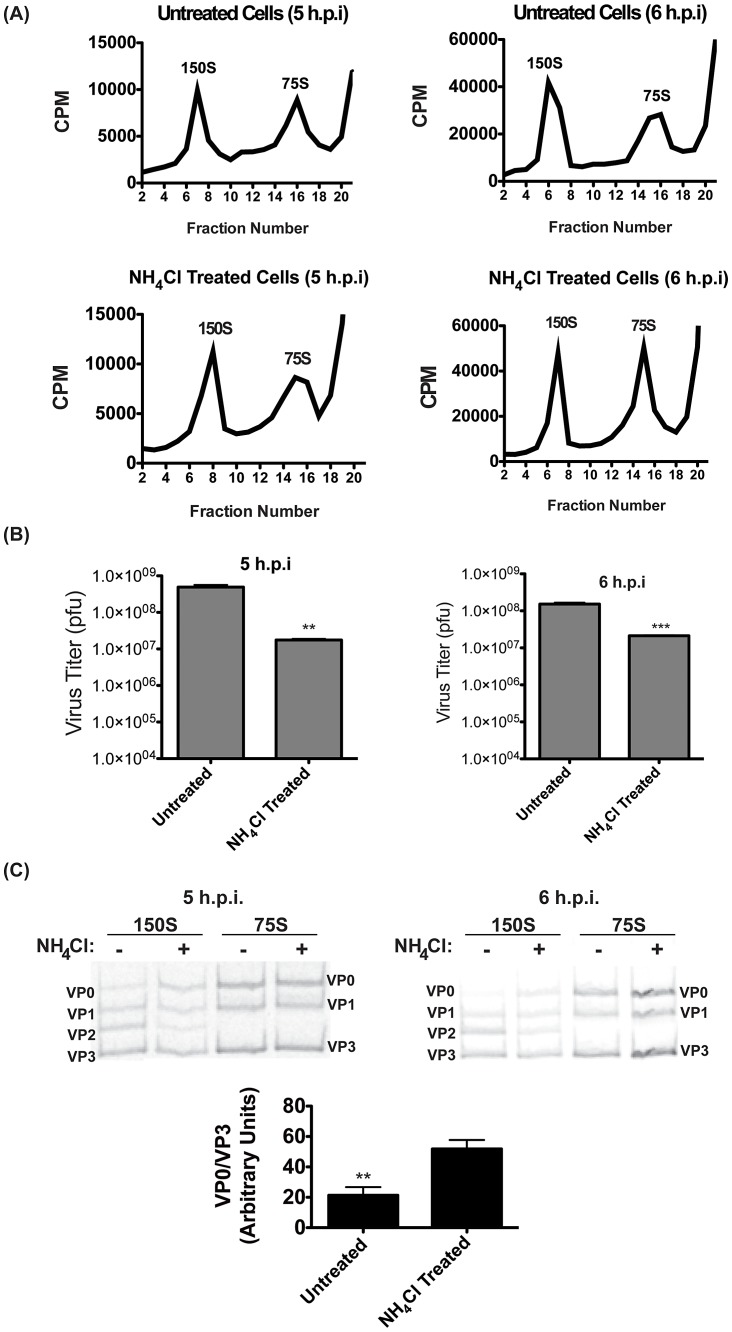
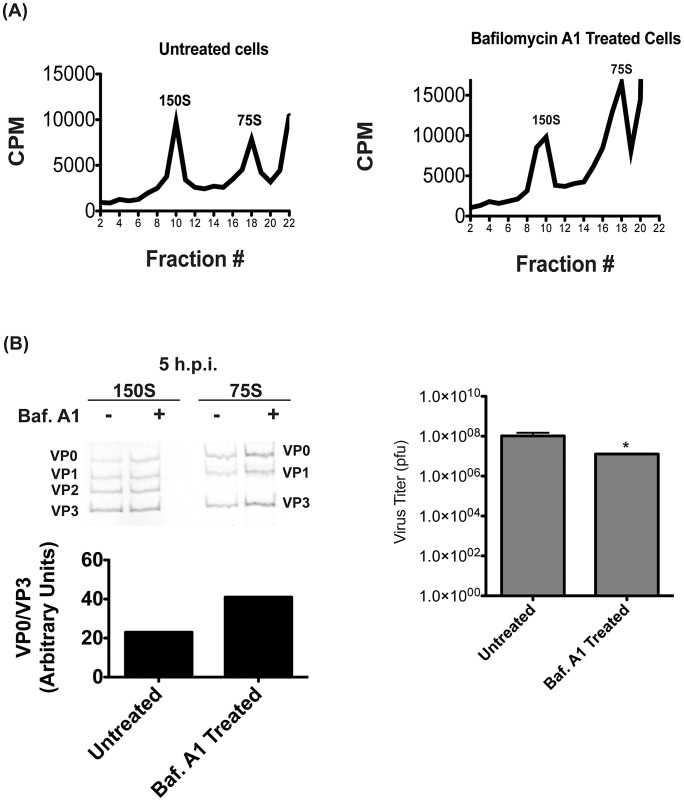
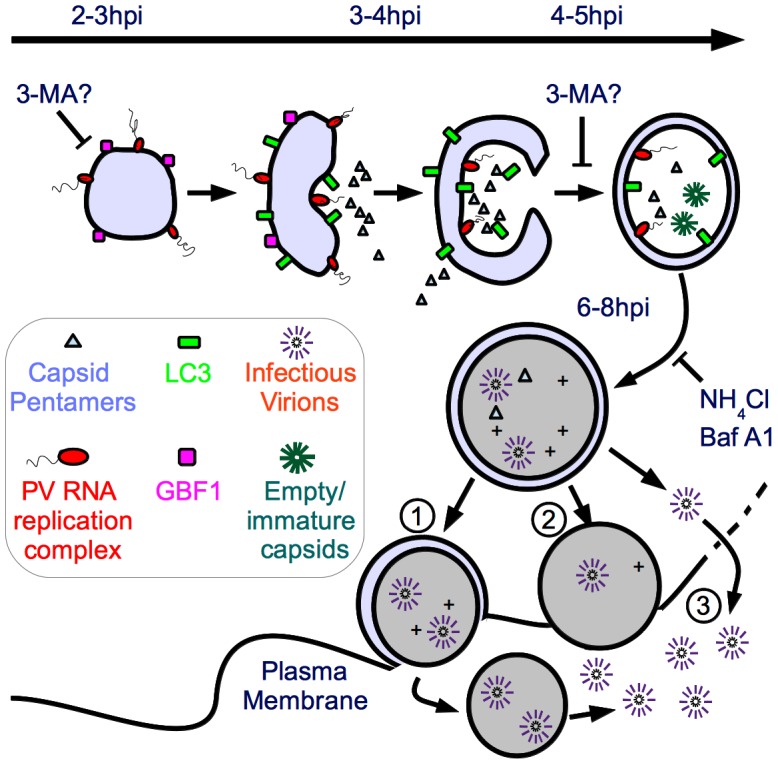
The autophagic pathway acts as part of the immune response against a variety of pathogens. However, several pathogens subvert autophagic signaling to promote their own replication. In many cases it has been demonstrated that these pathogens inhibit or delay the degradative aspect of autophagy. Here, using poliovirus as a model virus, we report for the first time bona fide autophagic degradation occurring during infection with a virus whose replication is promoted by autophagy. We found that this degradation is not required to promote poliovirus replication. However, vesicular acidification, which in the case of autophagy precedes delivery of cargo to lysosomes, is required for normal levels of virus production. We show that blocking autophagosome formation inhibits viral RNA synthesis and subsequent steps in the virus cycle, while inhibiting vesicle acidification only inhibits the final maturation cleavage of virus particles. We suggest that particle assembly, genome encapsidation, and virion maturation may occur in a cellular compartment, and we propose the acidic mature autophagosome as a candidate vesicle. We discuss the implications of our findings in understanding the late stages of poliovirus replication, including the formation and maturation of virions and egress of infectious virus from cells.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Viruses and arrested autophagosome development.Autophagy. 2009 Aug;5(6):870-1. doi: 10.4161/auto.9046. Epub 2009 Aug 19. Autophagy. 2009. PMID: 19502808
-
Role of microtubules in extracellular release of poliovirus.J Virol. 2009 Jul;83(13):6599-609. doi: 10.1128/JVI.01819-08. Epub 2009 Apr 15. J Virol. 2009. PMID: 19369338 Free PMC article.
-
That which does not degrade you makes you stronger: infectivity of poliovirus depends on vesicle acidification.Autophagy. 2013 May;9(5):806-7. doi: 10.4161/auto.23962. Epub 2013 Feb 25. Autophagy. 2013. PMID: 23439228 Free PMC article.
-
How positive-strand RNA viruses benefit from autophagosome maturation.J Virol. 2013 Sep;87(18):9966-72. doi: 10.1128/JVI.00460-13. Epub 2013 Jun 12. J Virol. 2013. PMID: 23760248 Free PMC article. Review.
-
Poliovirus assembly and encapsidation of genomic RNA.Adv Virus Res. 1996;46:1-68. Adv Virus Res. 1996. PMID: 8824697 Review. No abstract available.
Cited by
-
Positive-strand RNA virus replication organelles at a glance.J Cell Sci. 2024 Sep 1;137(17):jcs262164. doi: 10.1242/jcs.262164. Epub 2024 Sep 10. J Cell Sci. 2024. PMID: 39254430 Free PMC article. Review.
-
A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy.Cells. 2019 Jul 3;8(7):674. doi: 10.3390/cells8070674. Cells. 2019. PMID: 31277291 Free PMC article. Review.
-
SIRT-1 is required for release of enveloped enteroviruses.Elife. 2023 Oct 18;12:RP87993. doi: 10.7554/eLife.87993. Elife. 2023. PMID: 37850626 Free PMC article.
-
A dynamically evolving war between autophagy and pathogenic microorganisms.J Zhejiang Univ Sci B. 2022 Jan 15;23(1):19-41. doi: 10.1631/jzus.B2100285. J Zhejiang Univ Sci B. 2022. PMID: 35029086 Free PMC article.
-
Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit.Cell Rep. 2018 Mar 20;22(12):3304-3314. doi: 10.1016/j.celrep.2018.03.003. Cell Rep. 2018. PMID: 29562185 Free PMC article.
References
-
- Whitton JL, Cornell CT, Feuer R (2005) Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 3 10 765–776. - PubMed
-
- Dales S, Eggers HJ, Tamm I, Palade GE (1965) Electron microscopic study of the formation of poliovirus. Virology 26: 379–389. - PubMed
-
- Tanida I (2011) Autophagy basics. Microbiol Immunol 55 1 1–11. - PubMed
-
- Mizushima N (2007) Autophagy: Process and function. Genes Dev 21 22 2861–2873. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
