

Tinkering with translation: protein synthesis in virus-infected cells
- PMID: 23209131
- PMCID: PMC3579402
- DOI: 10.1101/cshperspect.a012351
Tinkering with translation: protein synthesis in virus-infected cells
Abstract
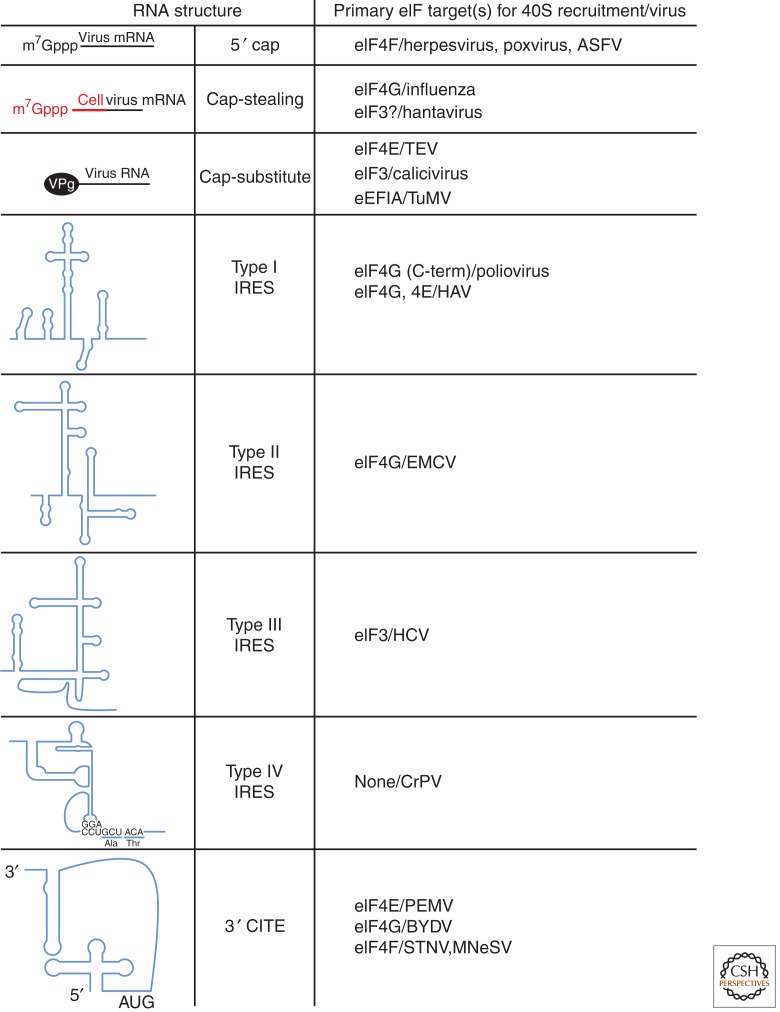
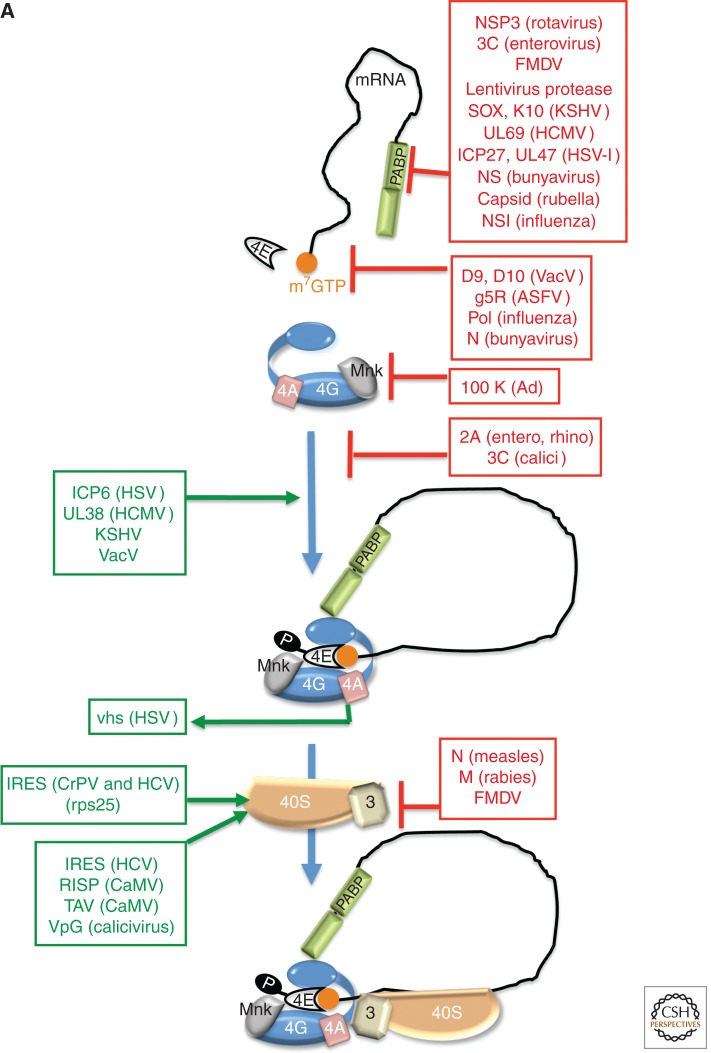
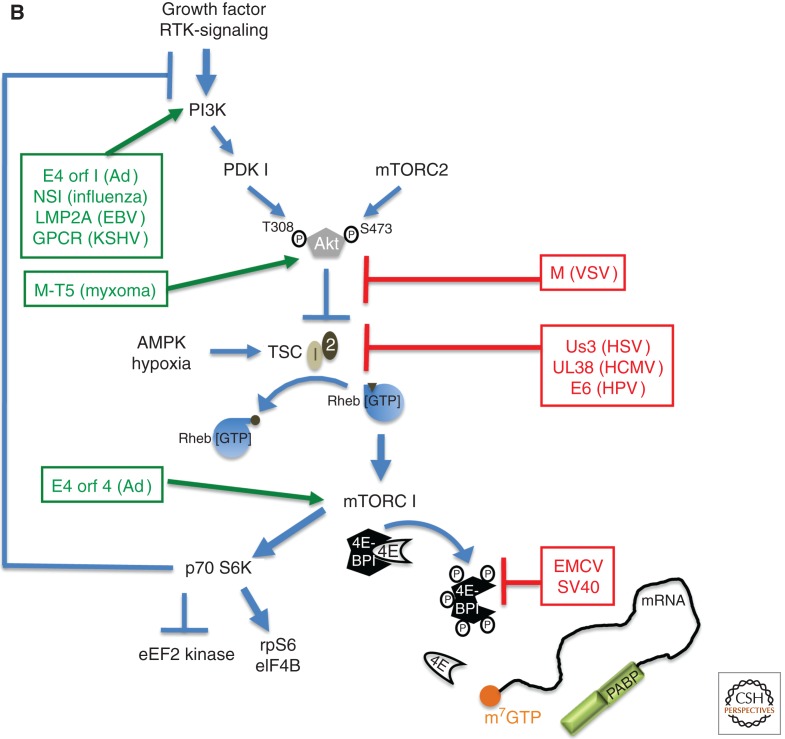
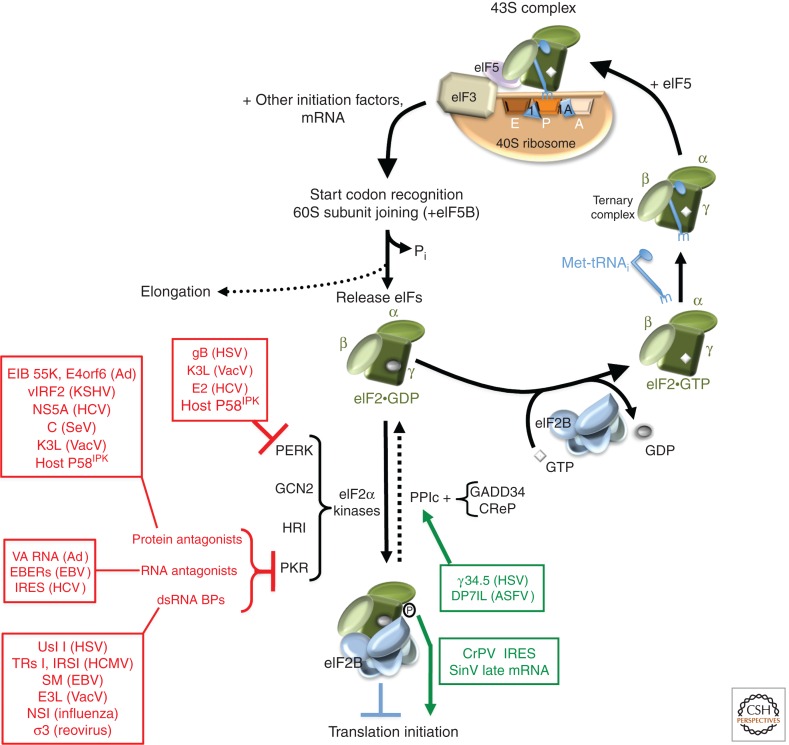
Viruses are obligate intracellular parasites, and their replication requires host cell functions. Although the size, composition, complexity, and functions encoded by their genomes are remarkably diverse, all viruses rely absolutely on the protein synthesis machinery of their host cells. Lacking their own translational apparatus, they must recruit cellular ribosomes in order to translate viral mRNAs and produce the protein products required for their replication. In addition, there are other constraints on viral protein production. Crucially, host innate defenses and stress responses capable of inactivating the translation machinery must be effectively neutralized. Furthermore, the limited coding capacity of the viral genome needs to be used optimally. These demands have resulted in complex interactions between virus and host that exploit ostensibly virus-specific mechanisms and, at the same time, illuminate the functioning of the cellular protein synthesis apparatus.
Figures
Similar articles
-
Viral subversion of the host protein synthesis machinery.Nat Rev Microbiol. 2011 Oct 17;9(12):860-75. doi: 10.1038/nrmicro2655. Nat Rev Microbiol. 2011. PMID: 22002165 Free PMC article. Review.
-
A Cap-to-Tail Guide to mRNA Translation Strategies in Virus-Infected Cells.Annu Rev Virol. 2016 Sep 29;3(1):283-307. doi: 10.1146/annurev-virology-100114-055014. Epub 2016 Aug 3. Annu Rev Virol. 2016. PMID: 27501262 Review.
-
Translational Control in Virus-Infected Cells.Cold Spring Harb Perspect Biol. 2019 Mar 1;11(3):a033001. doi: 10.1101/cshperspect.a033001. Cold Spring Harb Perspect Biol. 2019. PMID: 29891561 Free PMC article. Review.
-
Takeover of host ribosomes by divergent IRES elements.Biochem Soc Trans. 2005 Dec;33(Pt 6):1479-82. doi: 10.1042/BST0331479. Biochem Soc Trans. 2005. PMID: 16246150 Review.
-
Translation initiation and viral tricks.Trends Biochem Sci. 2003 Mar;28(3):130-6. doi: 10.1016/S0968-0004(03)00029-X. Trends Biochem Sci. 2003. PMID: 12633992 Review.
Cited by
-
Coronavirus takeover of host cell translation and intracellular antiviral response: a molecular perspective.EMBO J. 2024 Jan;43(2):151-167. doi: 10.1038/s44318-023-00019-8. Epub 2024 Jan 10. EMBO J. 2024. PMID: 38200146 Free PMC article. Review.
-
Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology.Viruses. 2016 May 12;8(5):124. doi: 10.3390/v8050124. Viruses. 2016. PMID: 27187445 Free PMC article. Review.
-
Norovirus infection results in eIF2α independent host translation shut-off and remodels the G3BP1 interactome evading stress granule formation.PLoS Pathog. 2020 Jan 6;16(1):e1008250. doi: 10.1371/journal.ppat.1008250. eCollection 2020 Jan. PLoS Pathog. 2020. PMID: 31905230 Free PMC article.
-
Translation acrobatics: how cancer cells exploit alternate modes of translational initiation.EMBO Rep. 2018 Oct;19(10):e45947. doi: 10.15252/embr.201845947. Epub 2018 Sep 17. EMBO Rep. 2018. PMID: 30224410 Free PMC article. Review.
-
The emerging role of miRNAs in the pathogenesis of COVID-19: Protective effects of nutraceutical polyphenolic compounds against SARS-CoV-2 infection.Int J Med Sci. 2022 Jul 18;19(8):1340-1356. doi: 10.7150/ijms.76168. eCollection 2022. Int J Med Sci. 2022. PMID: 35928726 Free PMC article. Review.
References
-
- Andino R, Boddeker N, Silvera D, Gamarnik AV 1999. Intracellular determinants of picornavirus replication. Trends Microbiol 7: 76–82 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources