

Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling
- PMID: 23204521
- PMCID: PMC3543026
- DOI: 10.1074/jbc.M112.431973
Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling
Abstract
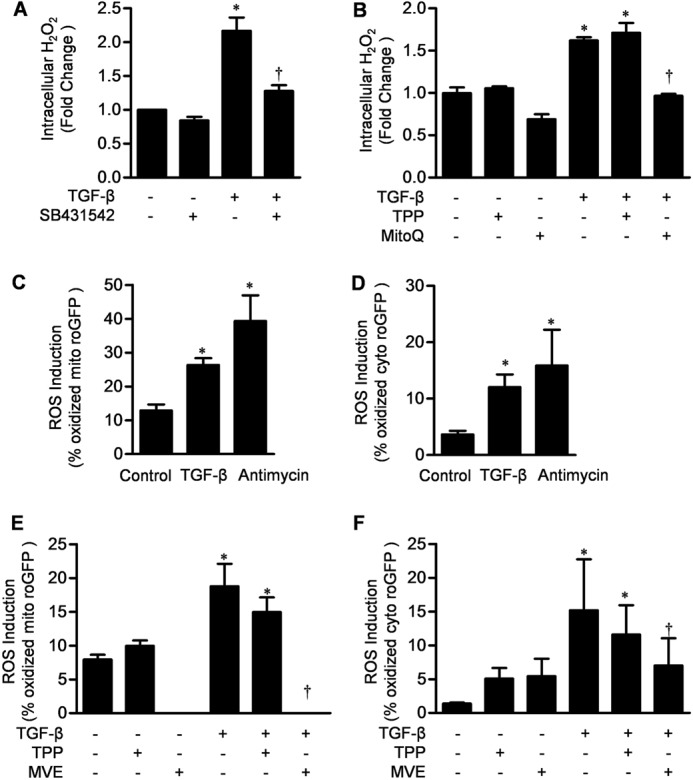
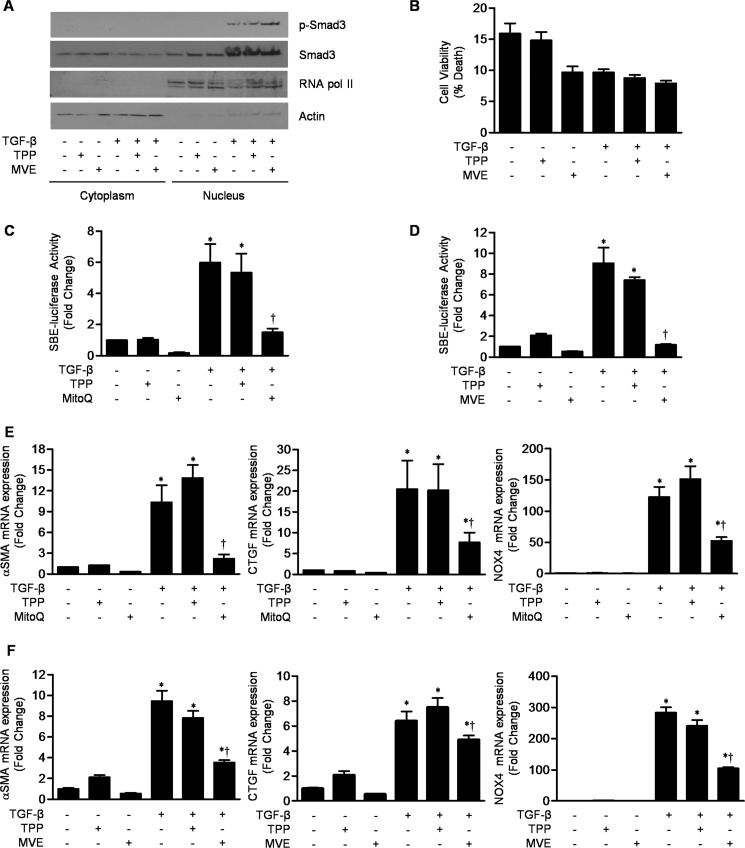
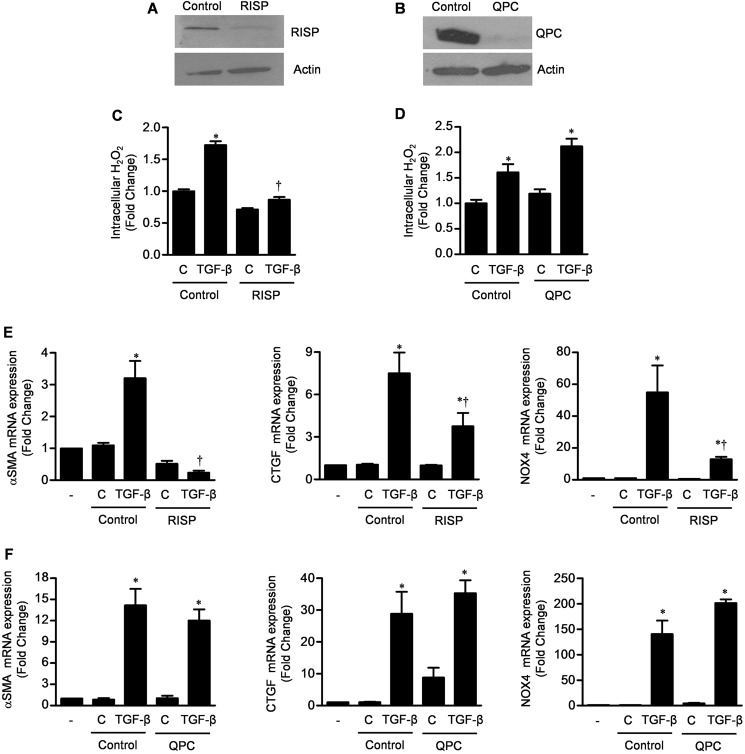
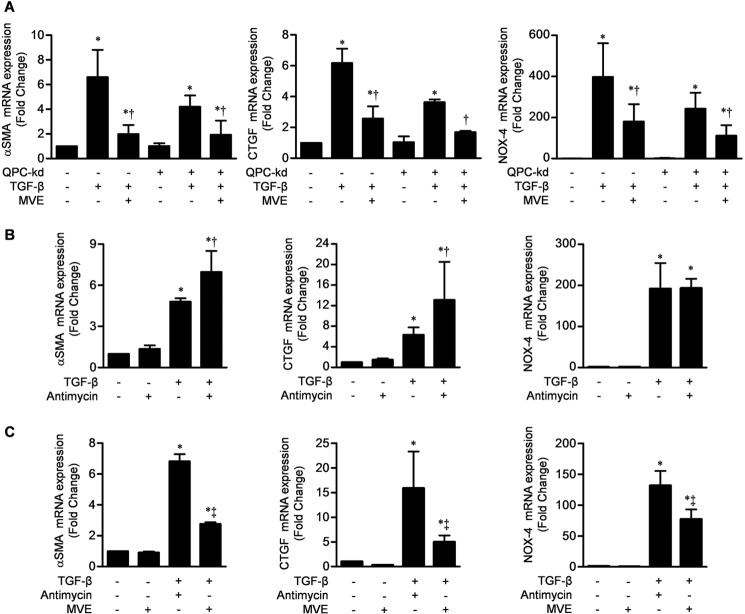
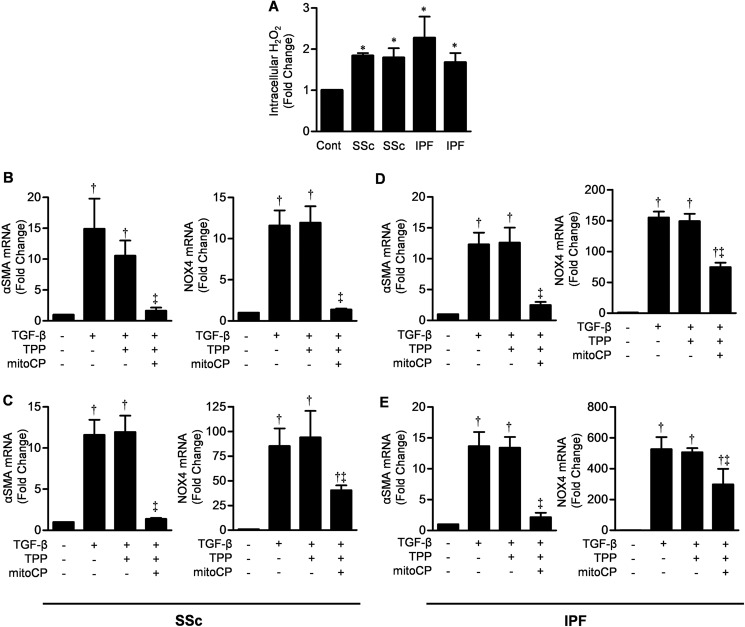
TGF-β signaling is required for normal tissue repair; however, excessive TGF-β signaling can lead to robust profibrotic gene expression in fibroblasts, resulting in tissue fibrosis. TGF-β binds to cell-surface receptors, resulting in the phosphorylation of the Smad family of transcription factors to initiate gene expression. TGF-β also initiates Smad-independent pathways, which augment gene expression. Here, we report that mitochondrial reactive oxygen species (ROS) generated at complex III are required for TGF-β-induced gene expression in primary normal human lung fibroblasts. TGF-β-induced ROS could be detected in both the mitochondrial matrix and cytosol. Mitochondrially targeted antioxidants markedly attenuated TGF-β-induced gene expression without affecting Smad phosphorylation or nuclear translocation. Genetically disrupting mitochondrial complex III-generated ROS production attenuated TGF-β-induced profibrotic gene expression. Furthermore, inhibiting mitochondrial ROS generation attenuated NOX4 (NADPH oxidase 4) expression, which is required for TGF-β induced myofibroblast differentiation. Lung fibroblasts from patients with pulmonary fibrosis generated more mitochondrial ROS than normal human lung fibroblasts, and mitochondrially targeted antioxidants attenuated profibrotic gene expression in both normal and fibrotic lung fibroblasts. Collectively, our results indicate that mitochondrial ROS are essential for normal TGF-β-mediated gene expression and that targeting mitochondrial ROS might be beneficial in diseases associated with excessive fibrosis.
Figures
Similar articles
-
Metformin attenuates lung fibrosis development via NOX4 suppression.Respir Res. 2016 Aug 30;17(1):107. doi: 10.1186/s12931-016-0420-x. Respir Res. 2016. PMID: 27576730 Free PMC article.
-
Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling.Proc Natl Acad Sci U S A. 2015 Apr 21;112(16):E2048-57. doi: 10.1073/pnas.1415111112. Epub 2015 Apr 6. Proc Natl Acad Sci U S A. 2015. PMID: 25848047 Free PMC article.
-
Profibrotic epithelial TGF-β1 signaling involves NOX4-mitochondria cross talk and redox-mediated activation of the tyrosine kinase FYN.Am J Physiol Lung Cell Mol Physiol. 2021 Mar 1;320(3):L356-L367. doi: 10.1152/ajplung.00444.2019. Epub 2020 Dec 16. Am J Physiol Lung Cell Mol Physiol. 2021. PMID: 33325804 Free PMC article.
-
TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities.Cell Signal. 2013 Jan;25(1):264-8. doi: 10.1016/j.cellsig.2012.10.003. Epub 2012 Oct 11. Cell Signal. 2013. PMID: 23063463 Free PMC article. Review.
-
Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis.Redox Biol. 2015 Dec;6:565-577. doi: 10.1016/j.redox.2015.09.009. Epub 2015 Oct 10. Redox Biol. 2015. PMID: 26496488 Free PMC article. Review.
Cited by
-
Immunomodulatory Effect of Green Tea Treatment in Combination with Low-dose Chemotherapy in Elderly Acute Myeloid Leukemia Patients with Myelodysplasia-related Changes.Integr Cancer Ther. 2021 Jan-Dec;20:15347354211002647. doi: 10.1177/15347354211002647. Integr Cancer Ther. 2021. PMID: 33754891 Free PMC article.
-
Mitochondrial DNA Sensing Pathogen Recognition Receptors in Systemic Sclerosis Associated Interstitial Lung Disease: A Review.Curr Treatm Opt Rheumatol. 2023 Dec;9(4):204-220. doi: 10.1007/s40674-023-00211-1. Epub 2023 Aug 8. Curr Treatm Opt Rheumatol. 2023. PMID: 38230363 Free PMC article.
-
3',4'-Dihydroxyflavonol Inhibits Fibrotic Response in a Rabbit Model of Glaucoma Filtration Surgery.Int J Mol Sci. 2024 Oct 7;25(19):10767. doi: 10.3390/ijms251910767. Int J Mol Sci. 2024. PMID: 39409096 Free PMC article.
-
From the Cover: Catalytic Antioxidant Rescue of Inhaled Sulfur Mustard Toxicity.Toxicol Sci. 2016 Dec;154(2):341-353. doi: 10.1093/toxsci/kfw170. Epub 2016 Sep 7. Toxicol Sci. 2016. PMID: 27605419 Free PMC article.
-
Adventitial Fibroblasts in Aortic Aneurysm: Unraveling Pathogenic Contributions to Vascular Disease.Diagnostics (Basel). 2022 Mar 31;12(4):871. doi: 10.3390/diagnostics12040871. Diagnostics (Basel). 2022. PMID: 35453919 Free PMC article. Review.
References
-
- Jennings M. T., Pietenpol J. A. (1998) The role of transforming growth factor β in glioma progression. J. Neurooncol. 36, 123–140 - PubMed
-
- Verrecchia F., Mauviel A. (2002) Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 118, 211–215 - PubMed
-
- Bhattacharyya S., Ghosh A. K., Pannu J., Mori Y., Takagawa S., Chen G., Trojanowska M., Gilliam A. C., Varga J. (2005) Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor β. Arthritis Rheum. 52, 1248–1258 - PubMed
-
- Hashimoto S., Gon Y., Takeshita I., Matsumoto K., Maruoka S., Horie T. (2001) Transforming growth factor-β1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun NH2-terminal kinase-dependent pathway. Am. J. Respir Crit. Care Med. 163, 152–157 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
