

Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation
- PMID: 23187627
- PMCID: PMC3514498
- DOI: 10.1038/ncomms2230
Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation
Abstract
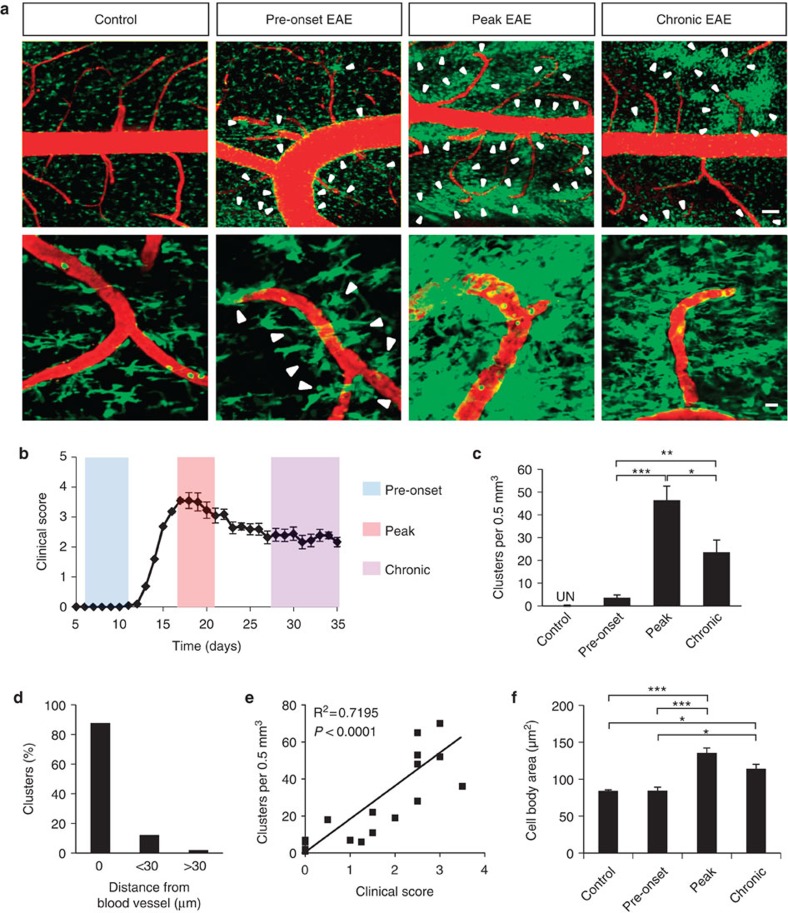
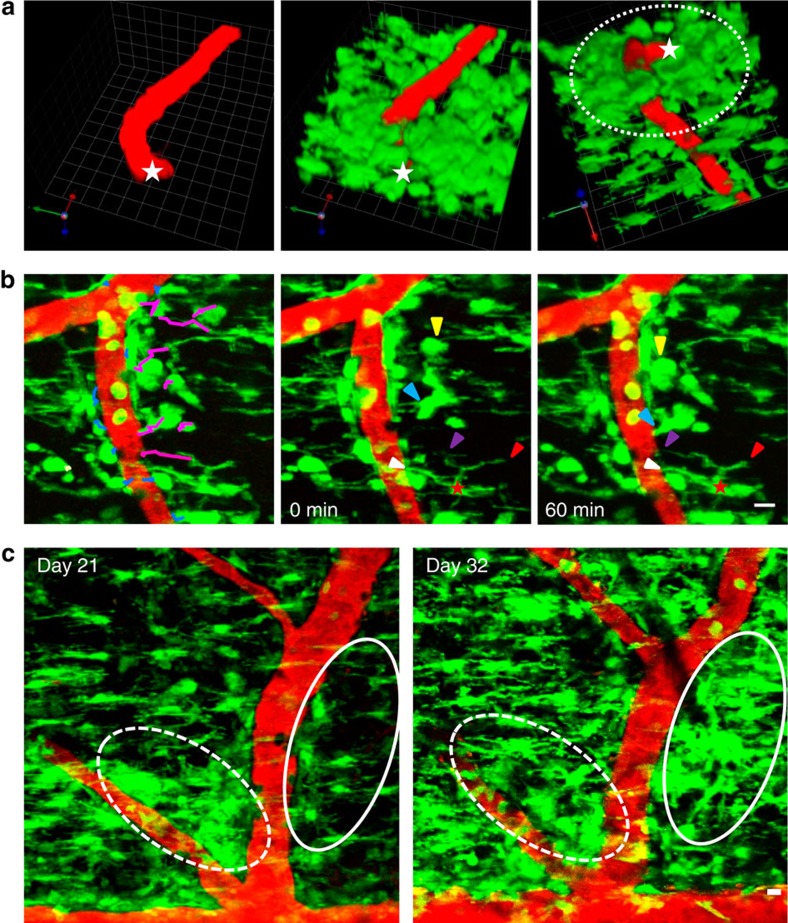
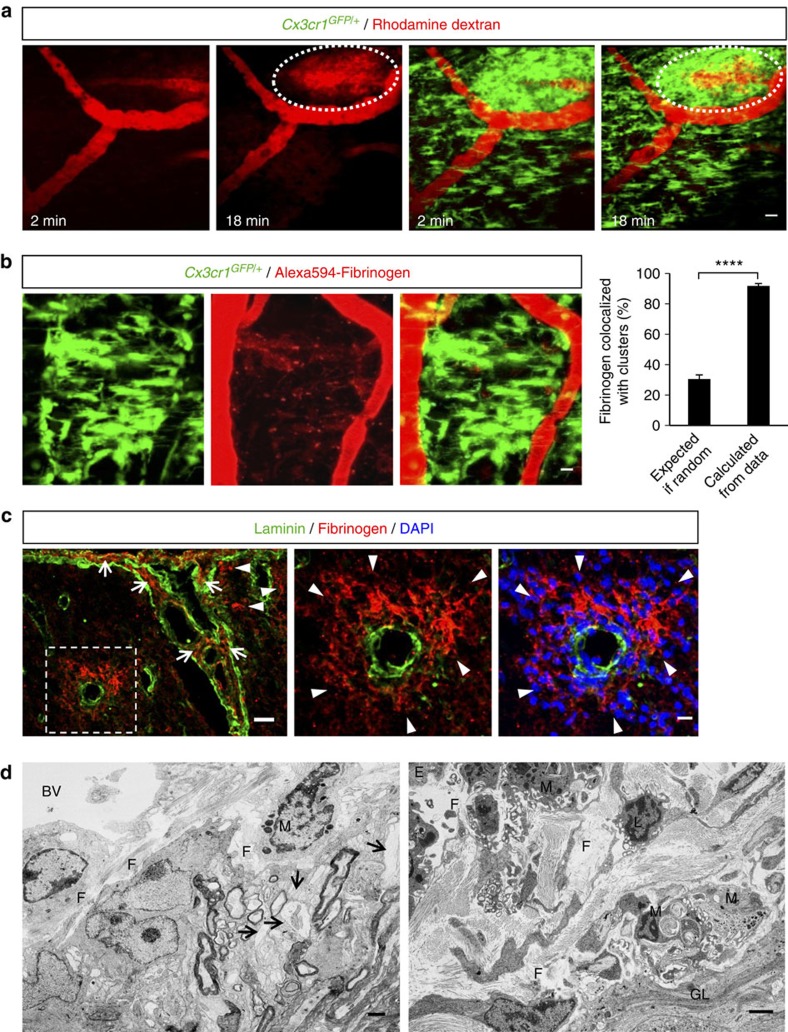
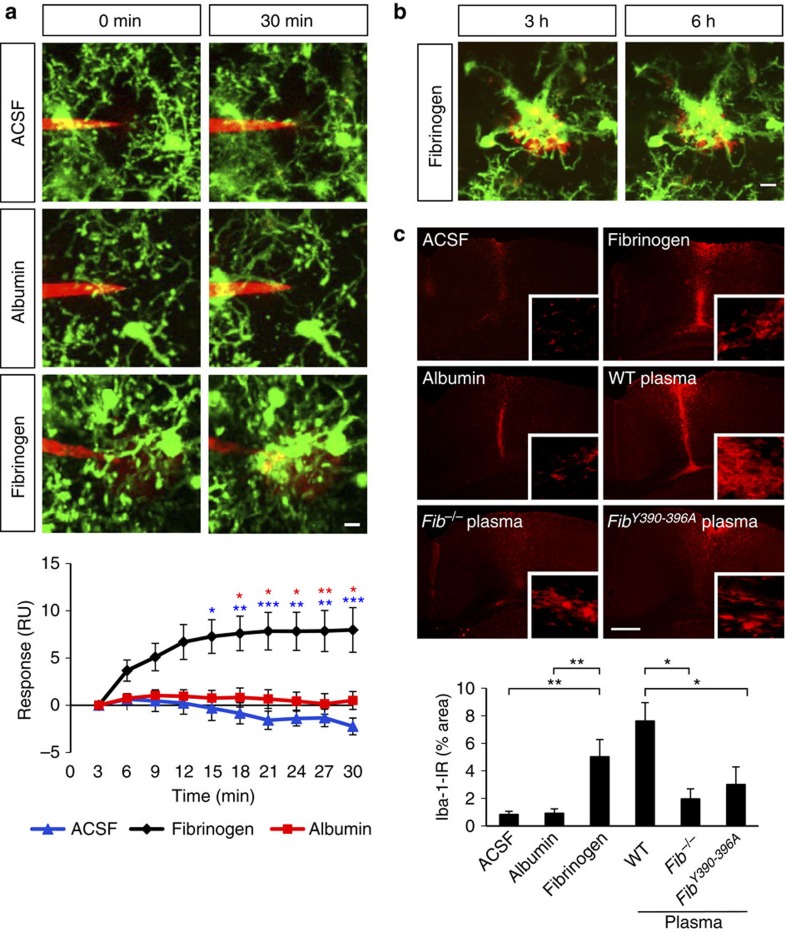
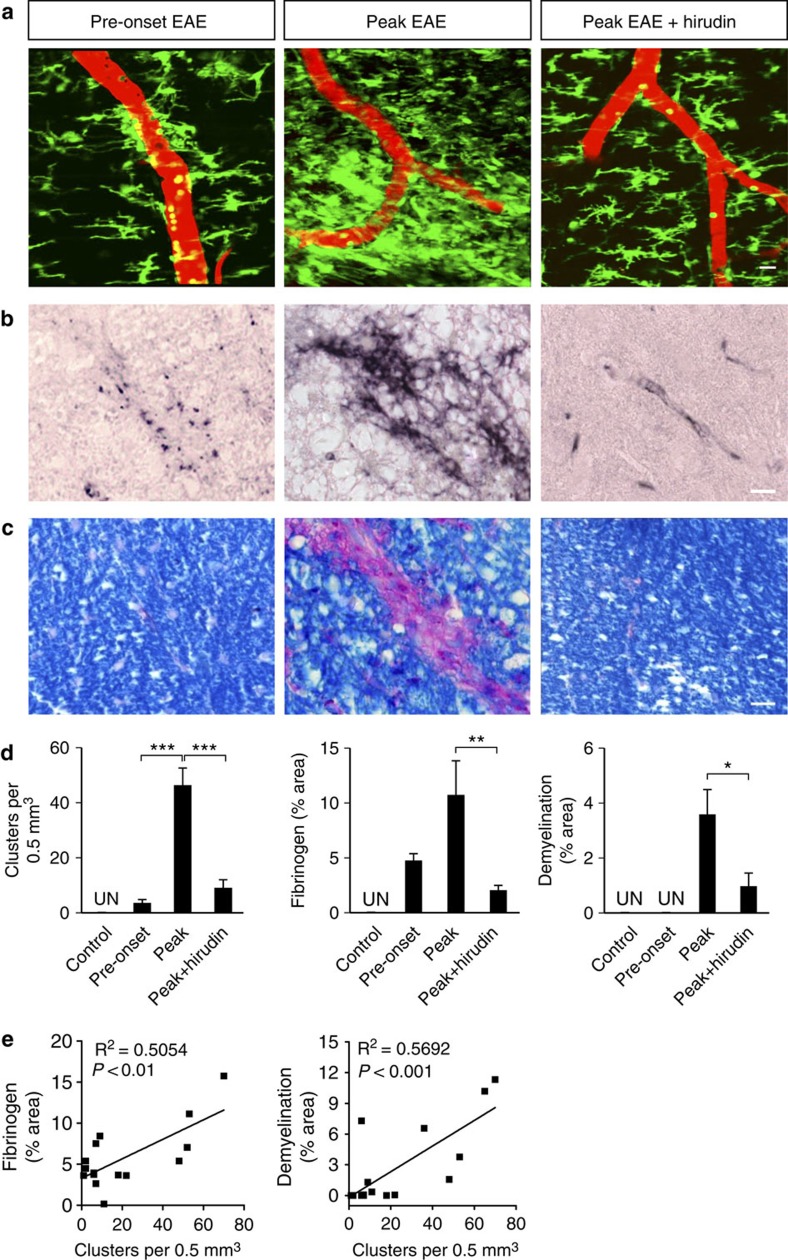
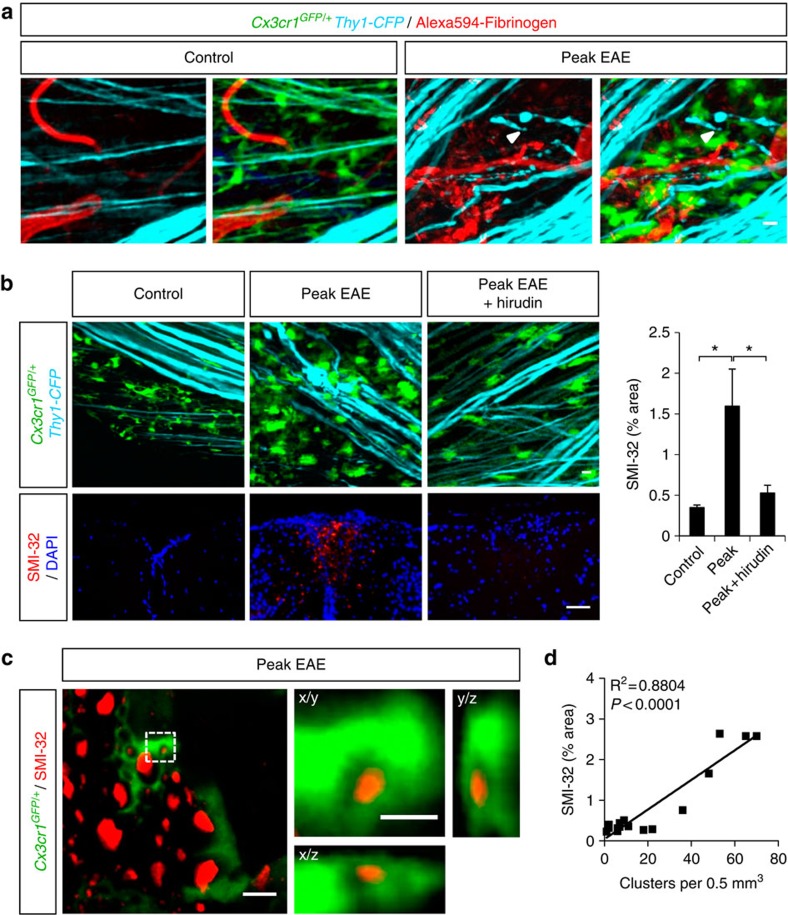
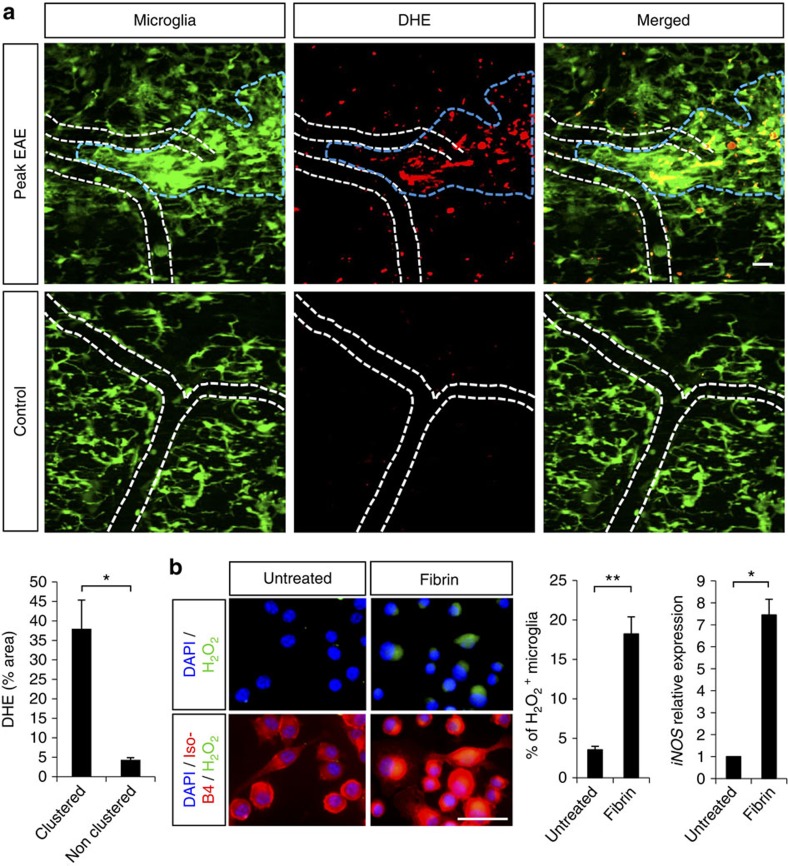
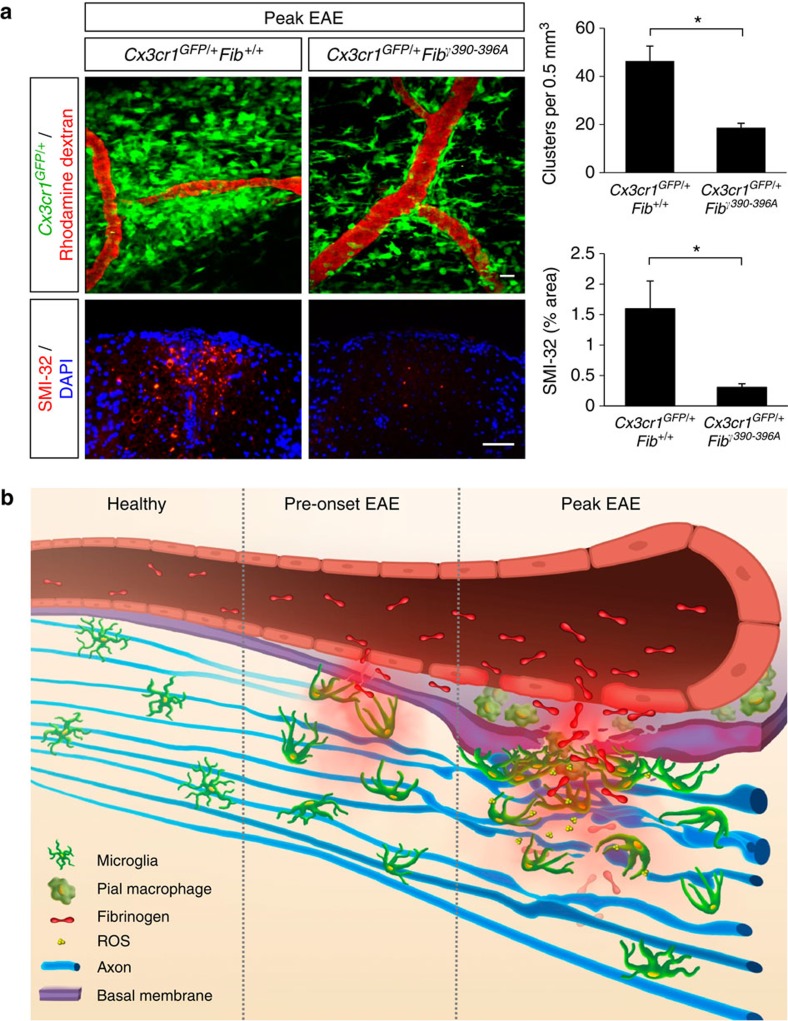
Blood-brain barrier disruption, microglial activation and neurodegeneration are hallmarks of multiple sclerosis. However, the initial triggers that activate innate immune responses and their role in axonal damage remain unknown. Here we show that the blood protein fibrinogen induces rapid microglial responses toward the vasculature and is required for axonal damage in neuroinflammation. Using in vivo two-photon microscopy, we demonstrate that microglia form perivascular clusters before myelin loss or paralysis onset and that, of the plasma proteins, fibrinogen specifically induces rapid and sustained microglial responses in vivo. Fibrinogen leakage correlates with areas of axonal damage and induces reactive oxygen species release in microglia. Blocking fibrin formation with anticoagulant treatment or genetically eliminating the fibrinogen binding motif recognized by the microglial integrin receptor CD11b/CD18 inhibits perivascular microglial clustering and axonal damage. Thus, early and progressive perivascular microglial clustering triggered by fibrinogen leakage upon blood-brain barrier disruption contributes to axonal damage in neuroinflammatory disease.
Conflict of interest statement
H. Lundbeck A/S is sponsoring research in Dr Akassoglou's laboratory to identify therapeutic candidates for neurological diseases. Dr Akassoglou is also a consultant for H. Lundbeck A/S.
Figures
Similar articles
-
The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease.J Exp Med. 2007 Mar 19;204(3):571-82. doi: 10.1084/jem.20061931. Epub 2007 Mar 5. J Exp Med. 2007. PMID: 17339406 Free PMC article.
-
Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer's Disease Model.Neuron. 2019 Mar 20;101(6):1099-1108.e6. doi: 10.1016/j.neuron.2019.01.014. Epub 2019 Feb 5. Neuron. 2019. PMID: 30737131 Free PMC article.
-
Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation.Nat Commun. 2015 Sep 10;6:8164. doi: 10.1038/ncomms9164. Nat Commun. 2015. PMID: 26353940 Free PMC article.
-
Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease.J Neurol Sci. 2003 Feb 15;206(2):165-71. doi: 10.1016/s0022-510x(02)00069-2. J Neurol Sci. 2003. PMID: 12559505 Review.
-
A Leaky Blood-Brain Barrier to Fibrinogen Contributes to Oxidative Damage in Alzheimer's Disease.Antioxidants (Basel). 2021 Dec 31;11(1):102. doi: 10.3390/antiox11010102. Antioxidants (Basel). 2021. PMID: 35052606 Free PMC article. Review.
Cited by
-
Engineering Brain-Specific Pericytes from Human Pluripotent Stem Cells.Tissue Eng Part B Rev. 2020 Aug;26(4):367-382. doi: 10.1089/ten.TEB.2020.0091. Tissue Eng Part B Rev. 2020. PMID: 32571167 Free PMC article. Review.
-
Ebola: translational science considerations.J Transl Med. 2015 Jan 16;13:11. doi: 10.1186/s12967-014-0362-3. J Transl Med. 2015. PMID: 25592846 Free PMC article. Review.
-
Assessing Microglial Dynamics by Live Imaging.Front Immunol. 2021 Mar 8;12:617564. doi: 10.3389/fimmu.2021.617564. eCollection 2021. Front Immunol. 2021. PMID: 33763064 Free PMC article. Review.
-
Dual microglia effects on blood brain barrier permeability induced by systemic inflammation.Nat Commun. 2019 Dec 20;10(1):5816. doi: 10.1038/s41467-019-13812-z. Nat Commun. 2019. PMID: 31862977 Free PMC article.
-
Glial and Vascular Cell Regulation of the Blood-Brain Barrier in Diabetes.Diabetes Metab J. 2022 Mar;46(2):222-238. doi: 10.4093/dmj.2021.0146. Epub 2022 Mar 18. Diabetes Metab J. 2022. PMID: 35299293 Free PMC article. Review.
References
-
- Lassmann H., Bruck W., Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends. Mol. Med. 7, 115–121 (2001). - PubMed
-
- Vos C. M. et al. Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol. Dis. 20, 953–960 (2005). - PubMed
-
- Garcia A. D., Doan N. B., Imura T., Bush T. G., Sofroniew M. V. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci. 7, 1233–1241 (2004). - PubMed
-
- van der Valk P., Amor S. Preactive lesions in multiple sclerosis. Curr. Opin. Neurol. 22, 207–213 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL096126/HL/NHLBI NIH HHS/United States
- NS052189/NS/NINDS NIH HHS/United States
- R01 NS066361/NS/NINDS NIH HHS/United States
- K12 HD000850/HD/NICHD NIH HHS/United States
- P41 RR004050/RR/NCRR NIH HHS/United States
- NS066361/NS/NINDS NIH HHS/United States
- CA082103/CA/NCI NIH HHS/United States
- NS051470/NS/NINDS NIH HHS/United States
- C06 RR018928/RR/NCRR NIH HHS/United States
- HL096126/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P30 CA082103/CA/NCI NIH HHS/United States
- RR18928/RR/NCRR NIH HHS/United States
- R01 NS052189/NS/NINDS NIH HHS/United States
- R01 NS051470/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
