

How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders?
- PMID: 23160421
- PMCID: PMC3923493
- DOI: 10.1097/WCO.0b013e32835a3efb
How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders?
Abstract
Purpose of review: The aim of this review is to describe disease mechanisms by which chromosome 9 open reading frame 72 (C9ORF72) repeat expansions could lead to amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) and to discuss these diseases in relation to other noncoding repeat expansion disorders.
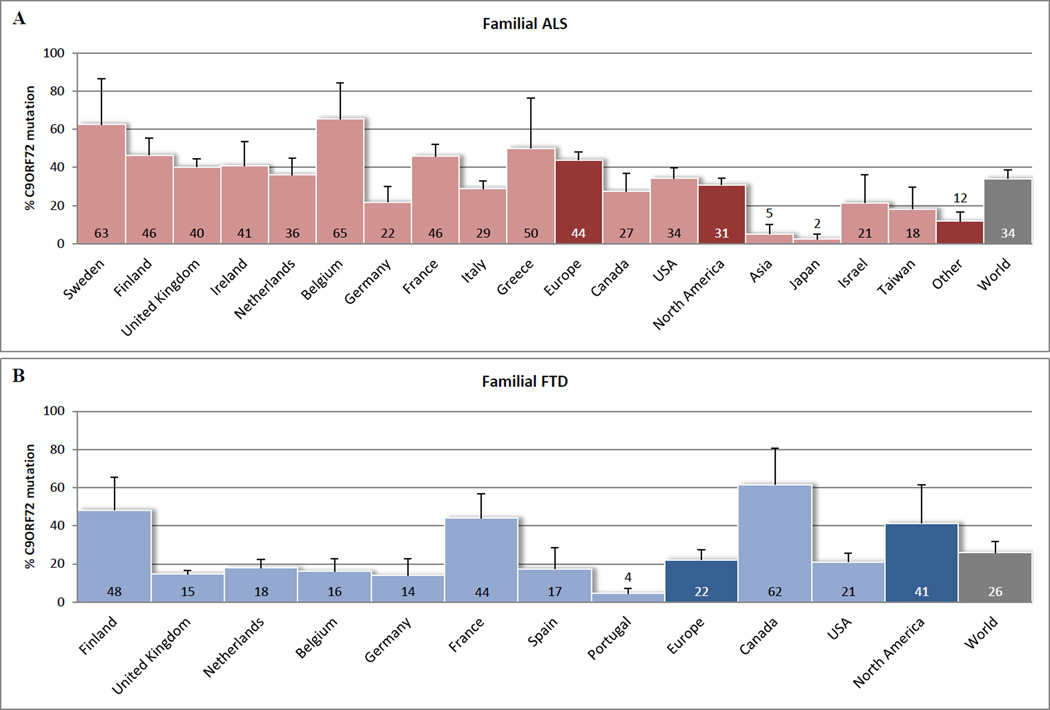
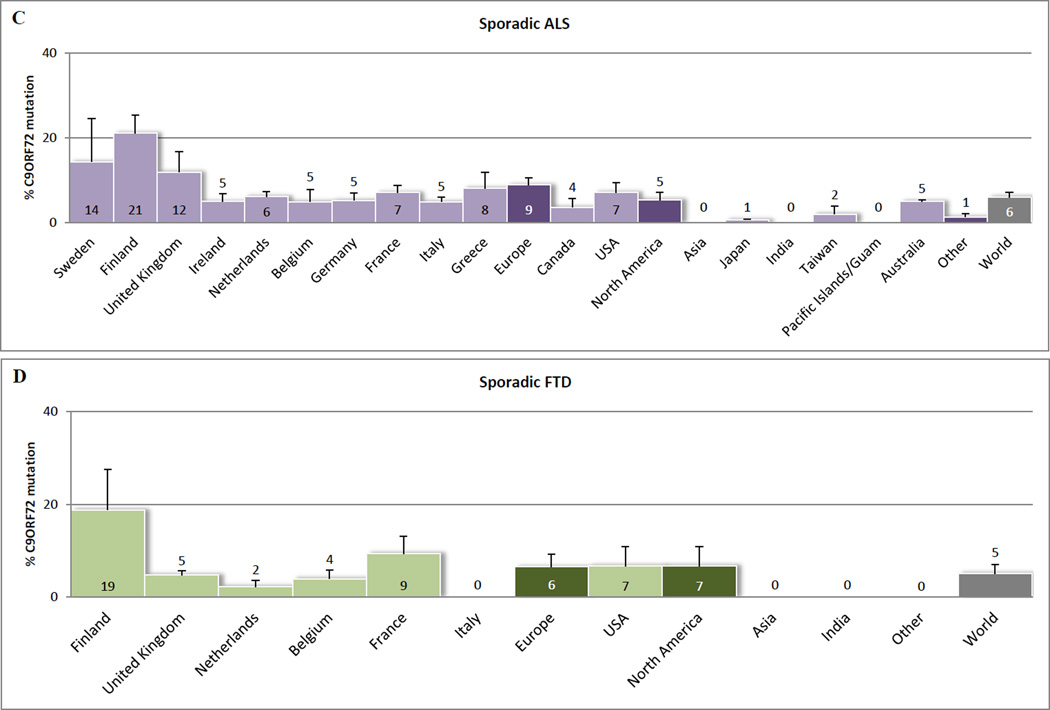
Recent findings: ALS and FTD are complex neurodegenerative disorders with a considerable clinical and pathological overlap, and this overlap is further substantiated by the recent discovery of C9ORF72 repeat expansions. These repeat expansions are currently the most important genetic cause of familial ALS and FTD, accounting for approximately 34.2 and 25.9% of the cases. Clinical phenotypes associated with these repeat expansions are highly variable, and combinations with mutations in other ALS-associated and/or FTD-associated genes may contribute to this pleiotropy. It is challenging, however, to diagnose patients with C9ORF72 expansions, not only because of large repeat sizes, but also due to somatic heterogeneity. Most other noncoding repeat expansion disorders share an RNA gain-of-function disease mechanism, a mechanism that could underlie the development of ALS and/or FTD as well.
Summary: The discovery of C9ORF72 repeat expansions provides novel insights into the pathogenesis of ALS and FTD and highlights the importance of noncoding repeat expansions and RNA toxicity in neurodegenerative diseases.
Conflict of interest statement
Disclosure statement: No actual or potential conflicts of interest have been reported.
Figures
Similar articles
-
Frontotemporal dementia with amyotrophic lateral sclerosis: a clinical comparison of patients with and without repeat expansions in C9orf72.Amyotroph Lateral Scler Frontotemporal Degener. 2013 Apr;14(3):172-6. doi: 10.3109/21678421.2013.765485. Epub 2013 Feb 19. Amyotroph Lateral Scler Frontotemporal Degener. 2013. PMID: 23421625
-
C9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels.Autophagy. 2021 Nov;17(11):3306-3322. doi: 10.1080/15548627.2021.1872189. Epub 2021 Feb 26. Autophagy. 2021. PMID: 33632058 Free PMC article. Review.
-
Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China.Neurobiol Aging. 2014 Apr;35(4):936.e19-22. doi: 10.1016/j.neurobiolaging.2013.10.001. Epub 2013 Oct 5. Neurobiol Aging. 2014. PMID: 24269022
-
Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype.Neurobiol Aging. 2012 Dec;33(12):2950.e5-7. doi: 10.1016/j.neurobiolaging.2012.07.005. Epub 2012 Jul 26. Neurobiol Aging. 2012. PMID: 22840558 Free PMC article.
-
Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions.J Neurochem. 2016 Aug;138 Suppl 1:145-62. doi: 10.1111/jnc.13623. Epub 2016 Jun 15. J Neurochem. 2016. PMID: 27016280 Review.
Cited by
-
Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS.Nat Neurosci. 2015 Aug;18(8):1175-82. doi: 10.1038/nn.4065. Epub 2015 Jul 20. Nat Neurosci. 2015. PMID: 26192745 Free PMC article.
-
Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes.Cells. 2022 Apr 1;11(7):1186. doi: 10.3390/cells11071186. Cells. 2022. PMID: 35406750 Free PMC article.
-
Frontotemporal dementia with a C9ORF72 expansion in a Swedish family: clinical and neuropathological characteristics.Am J Neurodegener Dis. 2013 Nov 29;2(4):276-86. eCollection 2013. Am J Neurodegener Dis. 2013. PMID: 24319645 Free PMC article.
-
The epidemiology of ALS: a conspiracy of genes, environment and time.Nat Rev Neurol. 2013 Nov;9(11):617-28. doi: 10.1038/nrneurol.2013.203. Epub 2013 Oct 15. Nat Rev Neurol. 2013. PMID: 24126629 Review.
-
GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport.Nature. 2015 Sep 3;525(7567):129-33. doi: 10.1038/nature14974. Epub 2015 Aug 26. Nature. 2015. PMID: 26308899 Free PMC article.
References
-
-
Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. This review summerizes the etiology of amyotrophic lateral sclerosis, its clinical characteristics, its diagnosis, its prognosis, and its management.
-
-
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27:48–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
