

VE-cadherin signaling induces EB3 phosphorylation to suppress microtubule growth and assemble adherens junctions
- PMID: 23159740
- PMCID: PMC3627495
- DOI: 10.1016/j.molcel.2012.10.011
VE-cadherin signaling induces EB3 phosphorylation to suppress microtubule growth and assemble adherens junctions
Abstract
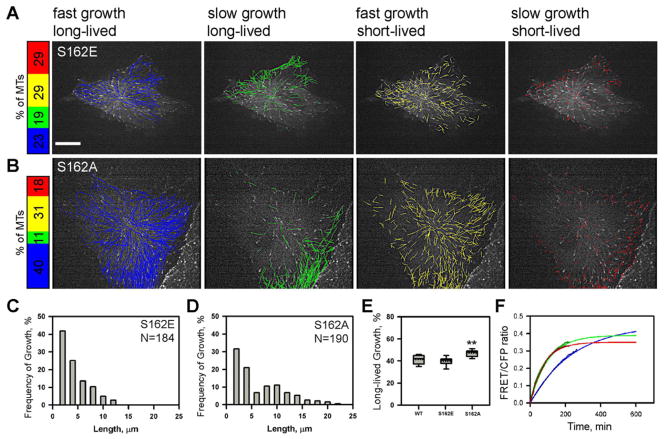
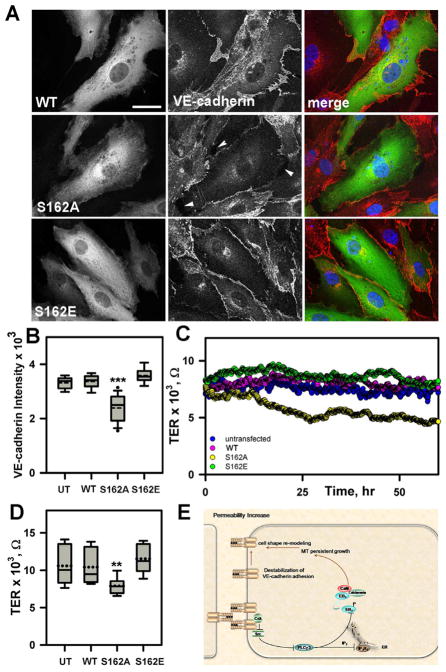
Vascular endothelial (VE)-cadherin homophilic adhesion controls endothelial barrier permeability through assembly of adherens junctions (AJs). We observed that loss of VE-cadherin-mediated adhesion induced the activation of Src and phospholipase C (PLC)γ2, which mediated Ca(2+) release from endoplasmic reticulum (ER) stores, resulting in activation of calcineurin (CaN), a Ca(2+)-dependent phosphatase. Downregulation of CaN activity induced phosphorylation of serine 162 in end binding (EB) protein 3. This phospho-switch was required to destabilize the EB3 dimer, suppress microtubule (MT) growth, and assemble AJs. The phospho-defective S162A EB3 mutant, in contrast, induced MT growth in confluent endothelial monolayers and disassembled AJs. Thus, VE-cadherin outside-in signaling regulates cytosolic Ca(2+) homeostasis and EB3 phosphorylation, which are required for assembly of AJs. These results identify a pivotal function of VE-cadherin homophilic interaction in modulating endothelial barrier through the tuning of MT dynamics.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
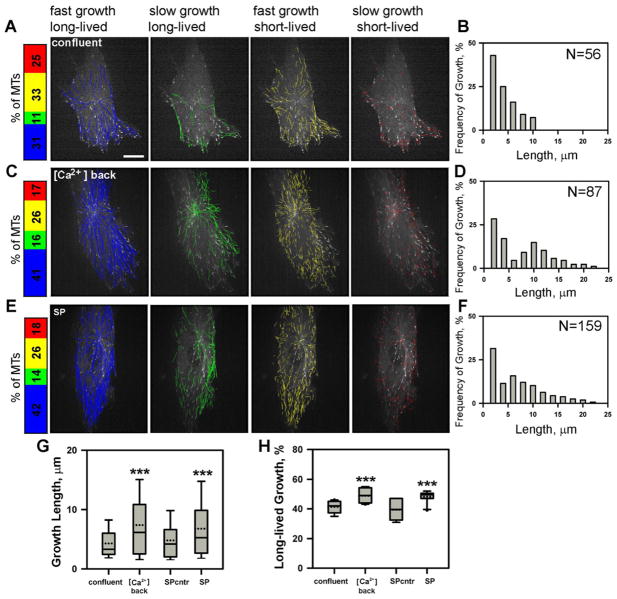
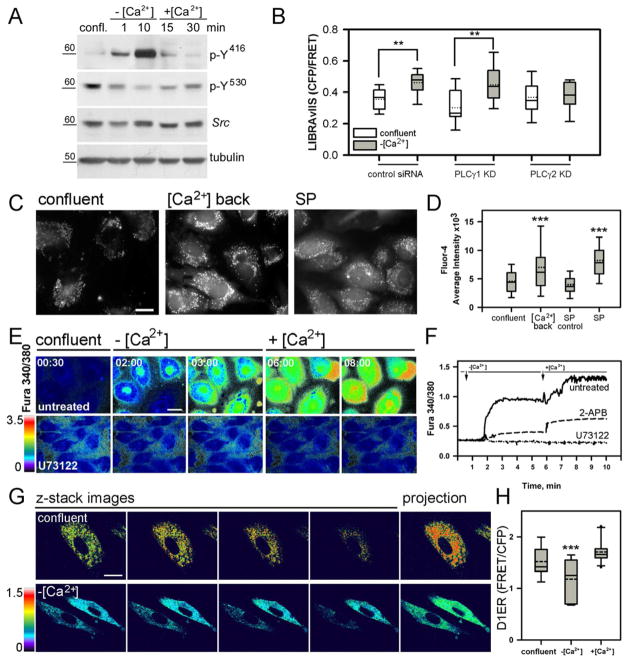
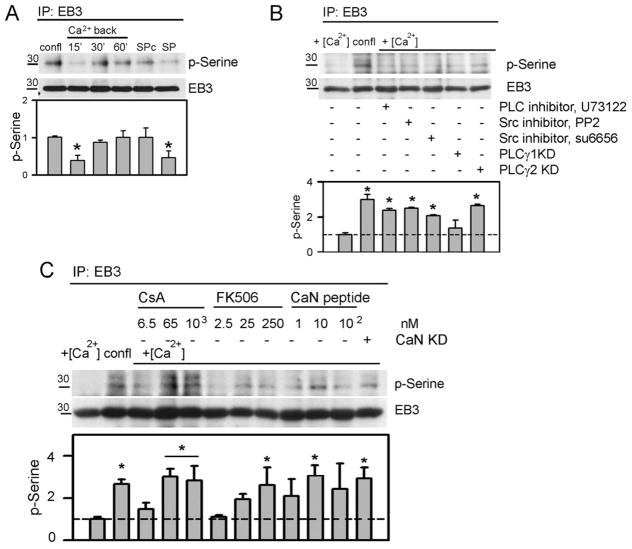
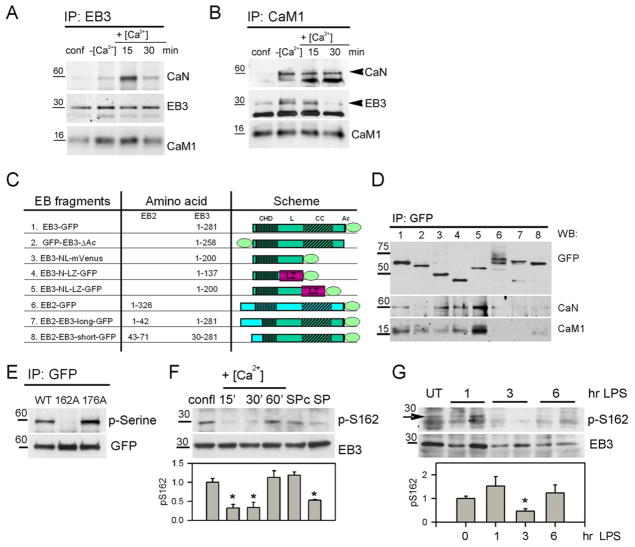
Similar articles
-
Pyk2 phosphorylation of VE-PTP downstream of STIM1-induced Ca2+ entry regulates disassembly of adherens junctions.Am J Physiol Lung Cell Mol Physiol. 2017 Jun 1;312(6):L1003-L1017. doi: 10.1152/ajplung.00008.2017. Epub 2017 Apr 6. Am J Physiol Lung Cell Mol Physiol. 2017. PMID: 28385807 Free PMC article.
-
Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers.J Biol Chem. 2010 Mar 5;285(10):7045-55. doi: 10.1074/jbc.M109.079277. Epub 2010 Jan 4. J Biol Chem. 2010. PMID: 20048167 Free PMC article.
-
PKCα activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity.Circ Res. 2012 Aug 31;111(6):739-49. doi: 10.1161/CIRCRESAHA.112.269654. Epub 2012 Jul 12. Circ Res. 2012. PMID: 22798526 Free PMC article.
-
The role of adherens junctions and VE-cadherin in the control of vascular permeability.J Cell Sci. 2008 Jul 1;121(Pt 13):2115-22. doi: 10.1242/jcs.017897. J Cell Sci. 2008. PMID: 18565824 Review.
-
Dynamic Regulation of Vascular Permeability by Vascular Endothelial Cadherin-Mediated Endothelial Cell-Cell Junctions.J Nippon Med Sch. 2017;84(4):148-159. doi: 10.1272/jnms.84.148. J Nippon Med Sch. 2017. PMID: 28978894 Review.
Cited by
-
VE-cadherin regulates migration inhibitory factor synthesis and release.Inflamm Res. 2019 Oct;68(10):877-887. doi: 10.1007/s00011-019-01270-8. Epub 2019 Jul 24. Inflamm Res. 2019. PMID: 31342095
-
LGN Directs Interphase Endothelial Cell Behavior via the Microtubule Network.PLoS One. 2015 Sep 23;10(9):e0138763. doi: 10.1371/journal.pone.0138763. eCollection 2015. PLoS One. 2015. PMID: 26398908 Free PMC article.
-
Transient receptor potential channel 1 maintains adherens junction plasticity by suppressing sphingosine kinase 1 expression to induce endothelial hyperpermeability.FASEB J. 2016 Jan;30(1):102-10. doi: 10.1096/fj.15-275891. Epub 2015 Aug 27. FASEB J. 2016. PMID: 26316271 Free PMC article.
-
Aurora B spatially regulates EB3 phosphorylation to coordinate daughter cell adhesion with cytokinesis.J Cell Biol. 2013 May 27;201(5):709-24. doi: 10.1083/jcb.201301131. J Cell Biol. 2013. PMID: 23712260 Free PMC article.
-
VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism.J Cell Biol. 2019 May 6;218(5):1725-1742. doi: 10.1083/jcb.201807210. Epub 2019 Apr 4. J Cell Biol. 2019. PMID: 30948425 Free PMC article.
References
-
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. The Journal of investigative dermatology. 1992;99:683–690. - PubMed
-
- Akhmanova A, Hoogenraad CC. Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol. 2005;17:47–54. - PubMed
-
- Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–322. - PubMed
-
- Bachmaier K, Toya S, Gao X, Triantafillou T, Garrean S, Park GY, Frey RS, Vogel S, Minshall R, Christman JW, et al. E3 ubiquitin ligase Cblb regulates the acute inflammatory response underlying lung injury. Nat Med. 2007;13:920–926. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
