

Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains
- PMID: 23152513
- PMCID: PMC3554088
- DOI: 10.1128/JVI.02517-12
Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains
Abstract
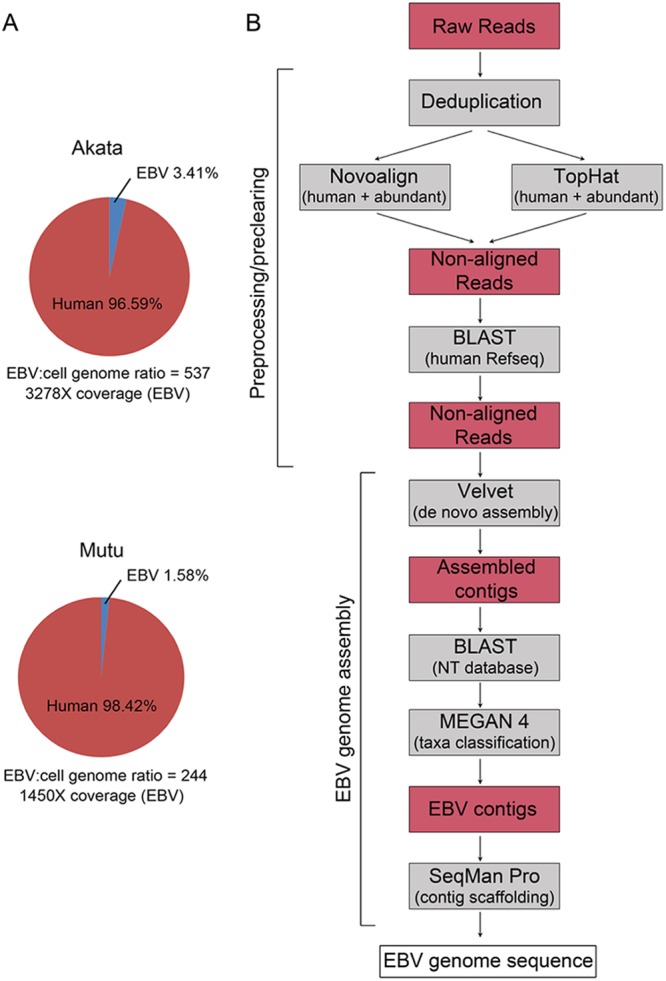
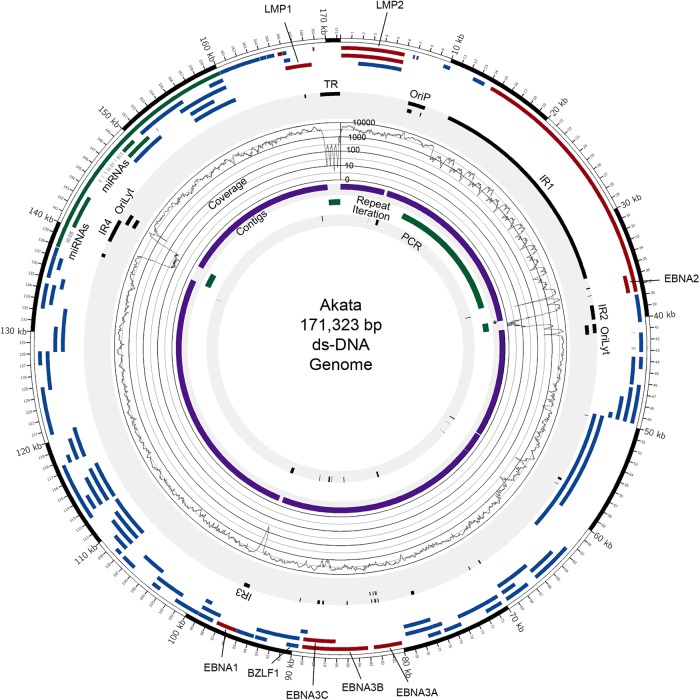
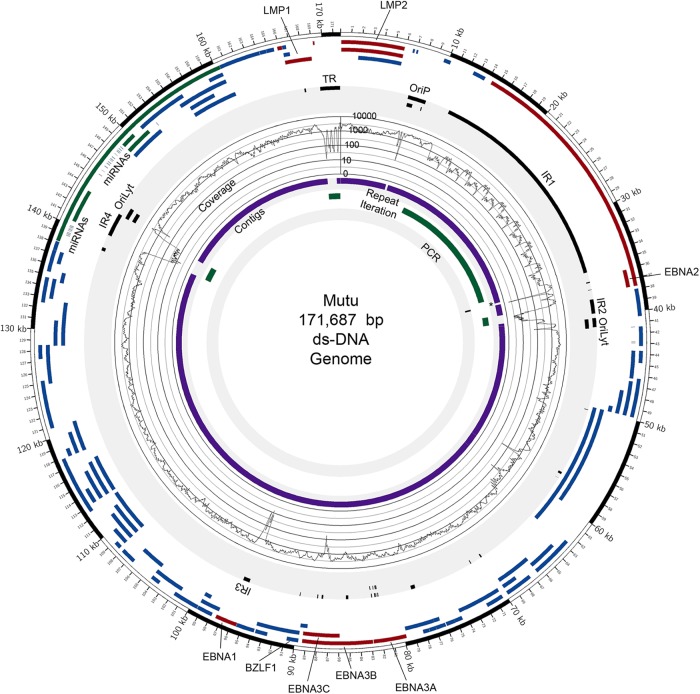
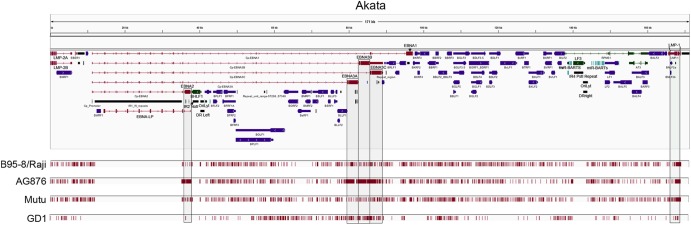
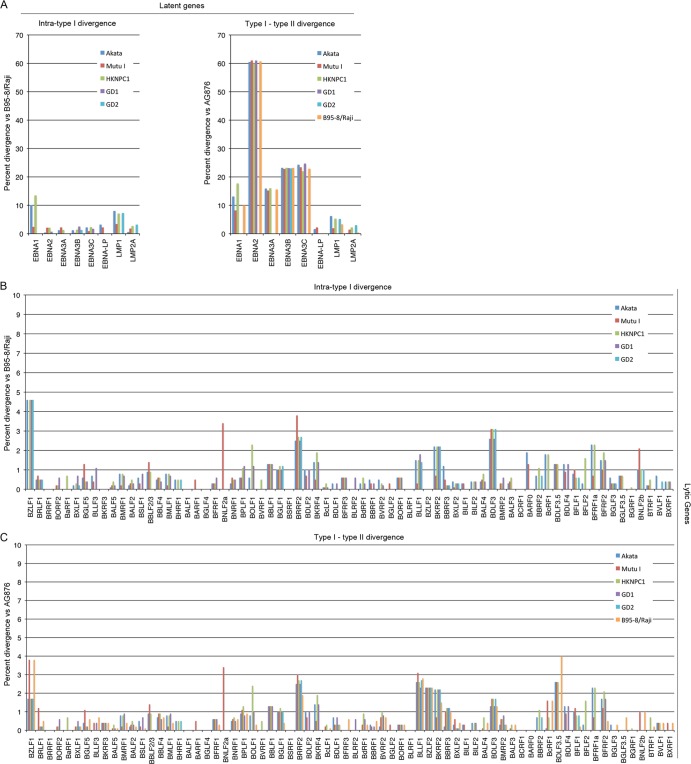
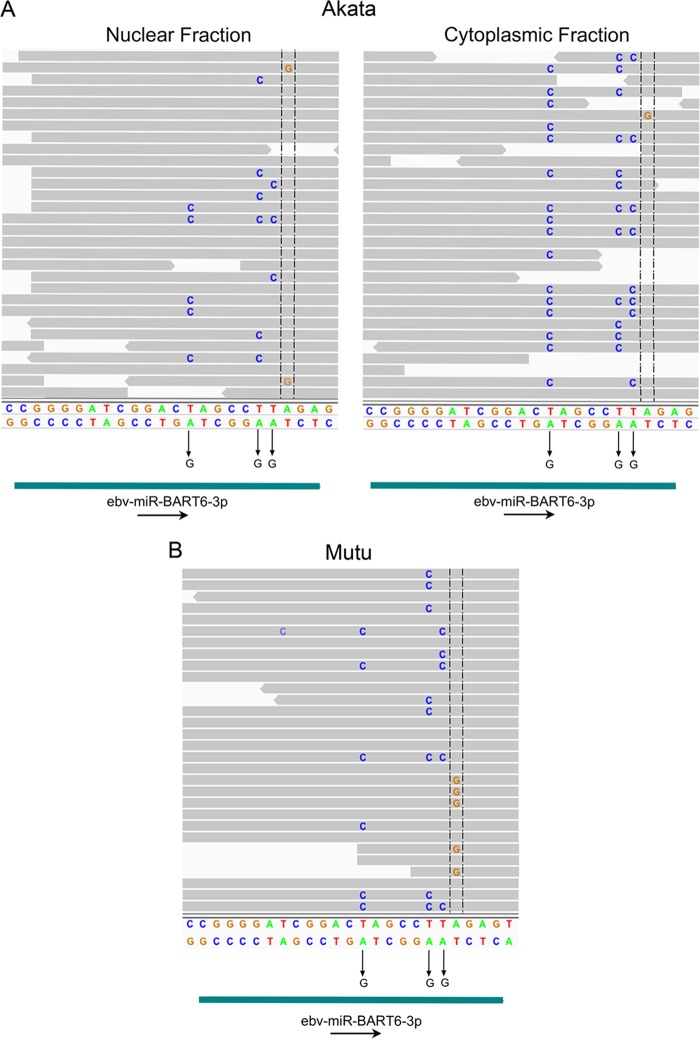
Using a simple viral genome enrichment approach, we report the de novo assembly of the Akata and Mutu Epstein-Barr virus (EBV) genomes from a single lane of next-generation sequencing (NGS) reads. The Akata and Mutu viral genomes are type I EBV strains of approximately 171 kb in length. Evidence for genome heterogeneity was found for the Akata but not for the Mutu strain. A comparative analysis of Akata with another four completely sequenced EBV strains, B95-8/Raji, AG876, Mutu, and GD1, demonstrated that the Akata strain is most closely related to the GD1 strain and exhibits the greatest divergence from the type II strain, AG876. A global comparison of latent and lytic gene sequences showed that the four latency genes, EBNA2, EBNA3A, EBNA3B, and EBNA3C, are uniquely defining of type I and type II strain differences. Within type I strains, LMP1, the latency gene, is among the most divergent of all EBV genes, with three insertion or deletion loci in its CTAR2 and CTAR3 signaling domains. Analysis of the BHLF1 and LF3 genes showed that the reading frames identified in the B95-8/Raji genome are not conserved in Akata (or Mutu, for BHLF1), suggesting a primarily non-protein-coding function in EBV's life cycle. The Akata and Mutu viral-genome sequences should be a useful resource for homology-based functional prediction and for molecular studies, such as PCR, RNA-seq, recombineering, and transcriptome studies. As an illustration, we identified novel RNA-editing events in ebv-miR-BART6 antisense transcripts using the Akata and Mutu reference genomes.
Figures



Similar articles
-
Differential regulation of Epstein-Barr virus (EBV) latent gene expression in Burkitt lymphoma cells infected with a recombinant EBV strain.J Virol. 2001 May;75(10):4929-35. doi: 10.1128/JVI.75.10.4929-4935.2001. J Virol. 2001. PMID: 11312367 Free PMC article.
-
Genomic sequencing and comparative analysis of Epstein-Barr virus genome isolated from primary nasopharyngeal carcinoma biopsy.PLoS One. 2012;7(5):e36939. doi: 10.1371/journal.pone.0036939. Epub 2012 May 10. PLoS One. 2012. PMID: 22590638 Free PMC article. Clinical Trial.
-
Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency.Virol J. 2017 Nov 13;14(1):221. doi: 10.1186/s12985-017-0891-5. Virol J. 2017. PMID: 29132393 Free PMC article.
-
Phylogenetic comparison of Epstein-Barr virus genomes.J Microbiol. 2018 Aug;56(8):525-533. doi: 10.1007/s12275-018-8039-x. Epub 2018 Jun 14. J Microbiol. 2018. PMID: 29948828 Review.
-
From Conventional to Next Generation Sequencing of Epstein-Barr Virus Genomes.Viruses. 2016 Feb 24;8(3):60. doi: 10.3390/v8030060. Viruses. 2016. PMID: 26927157 Free PMC article. Review.
Cited by
-
Tales from the crypt and coral reef: the successes and challenges of identifying new herpesviruses using metagenomics.Front Microbiol. 2015 Mar 13;6:188. doi: 10.3389/fmicb.2015.00188. eCollection 2015. Front Microbiol. 2015. PMID: 25821447 Free PMC article.
-
Single-Cell Analysis Reveals Malignant Cells Reshape the Cellular Landscape and Foster an Immunosuppressive Microenvironment of Extranodal NK/T-Cell Lymphoma.Adv Sci (Weinh). 2023 Dec;10(36):e2303913. doi: 10.1002/advs.202303913. Epub 2023 Nov 10. Adv Sci (Weinh). 2023. PMID: 37949673 Free PMC article.
-
Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection.J Virol. 2015 May;89(10):5222-37. doi: 10.1128/JVI.03614-14. Epub 2015 Mar 18. J Virol. 2015. PMID: 25787276 Free PMC article.
-
Epstein-Barr virus strain variation and cancer.Cancer Sci. 2019 Apr;110(4):1132-1139. doi: 10.1111/cas.13954. Epub 2019 Feb 21. Cancer Sci. 2019. PMID: 30697862 Free PMC article. Review.
-
Structural and Functional Basis for an EBNA1 Hexameric Ring in Epstein-Barr Virus Episome Maintenance.J Virol. 2017 Sep 12;91(19):e01046-17. doi: 10.1128/JVI.01046-17. Print 2017 Oct 1. J Virol. 2017. PMID: 28701406 Free PMC article.
References
-
- Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA
-
- Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA
-
- Takada K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27–32 - PubMed
-
- Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211 - PubMed
-
- Parker BD, Bankier A, Satchwell S, Barrell B, Farrell PJ. 1990. Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179:339–346 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
