

Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation
- PMID: 23149447
- PMCID: PMC3565767
- DOI: 10.1038/tp.2012.109
Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation
Abstract
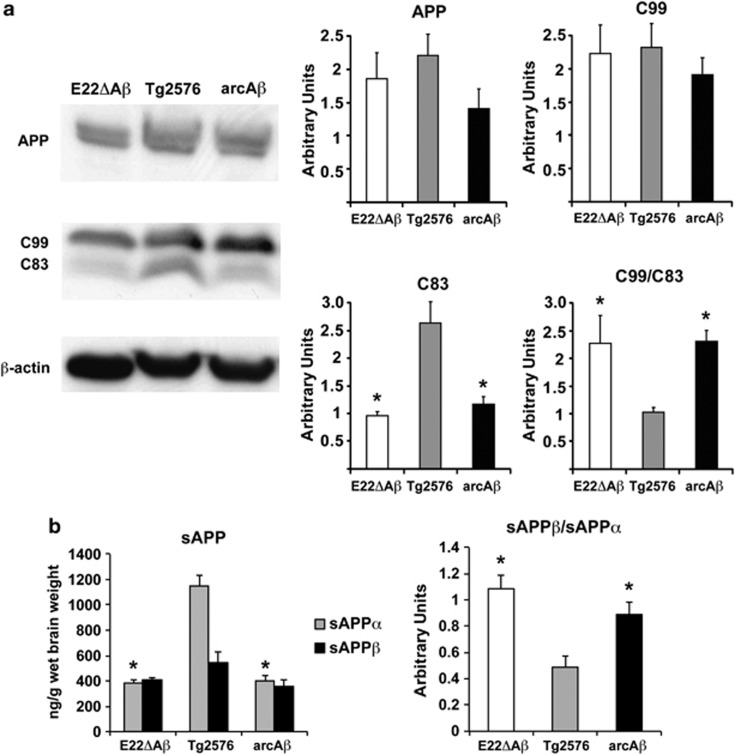
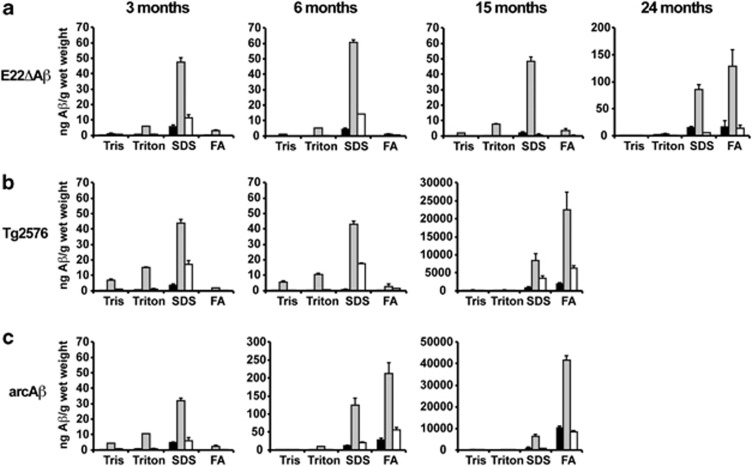
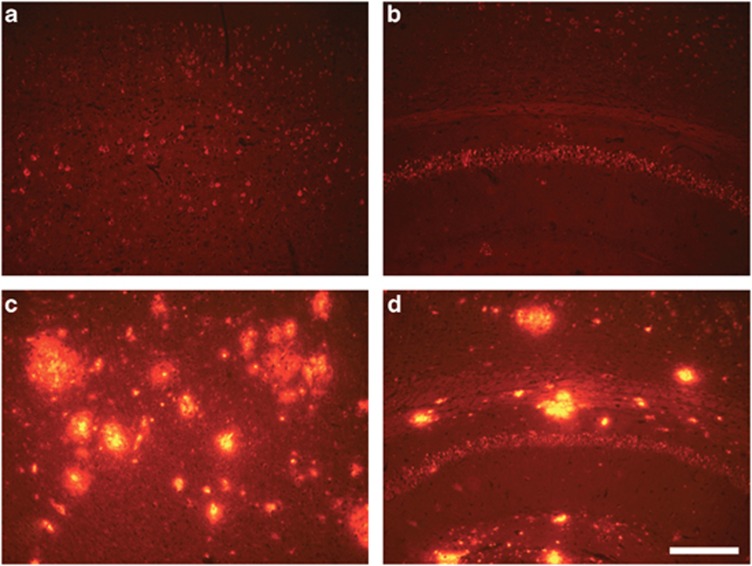
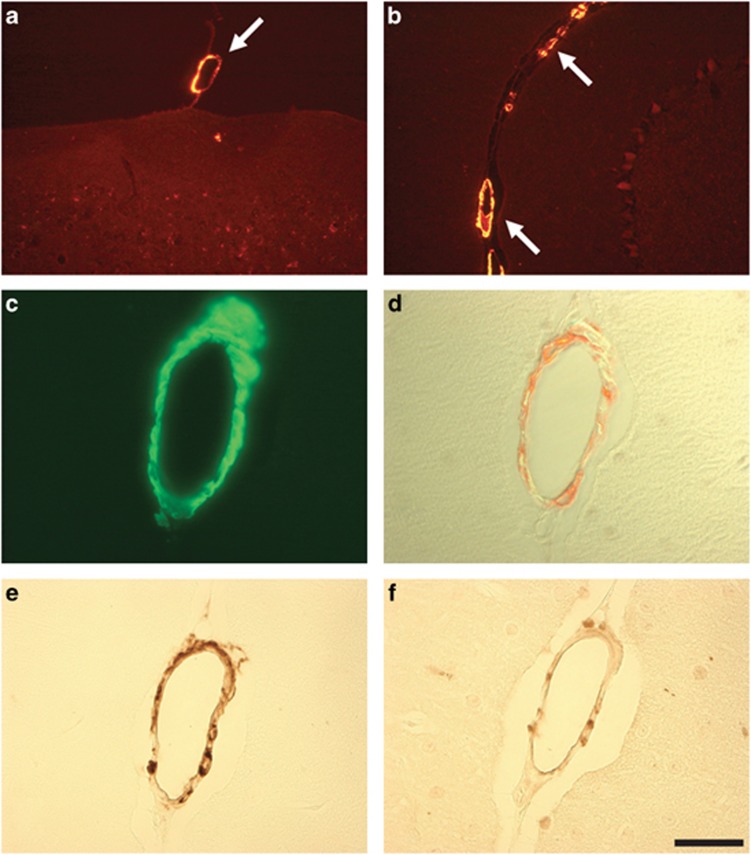
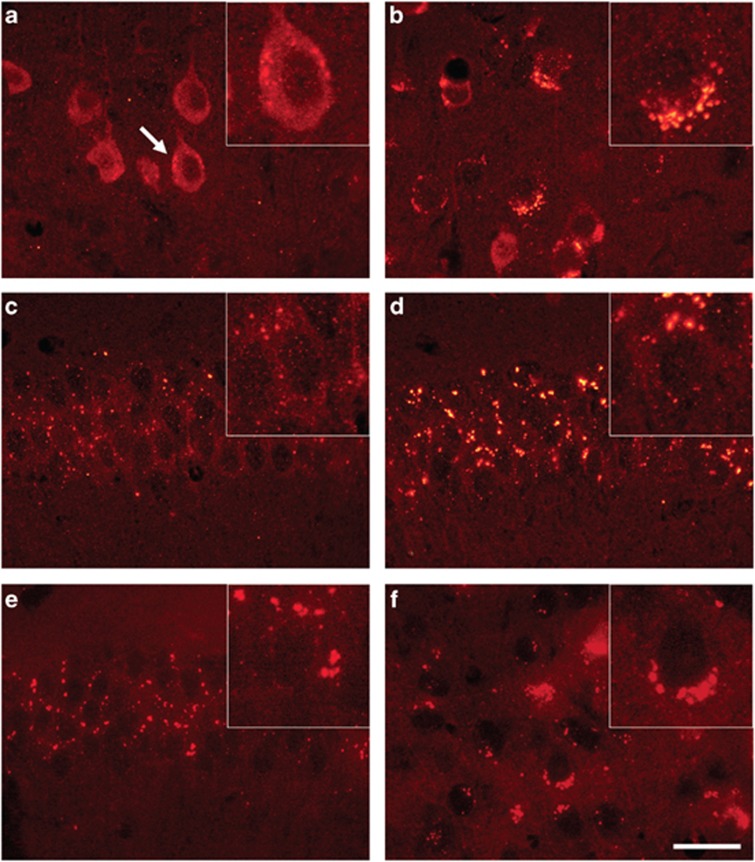
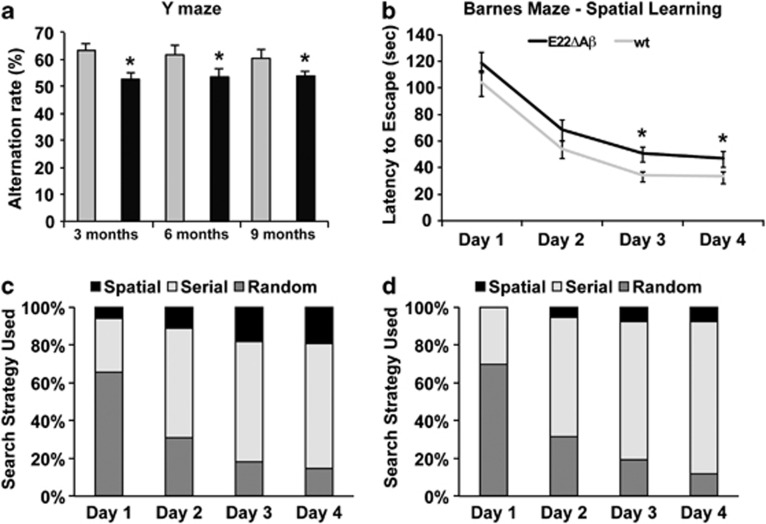
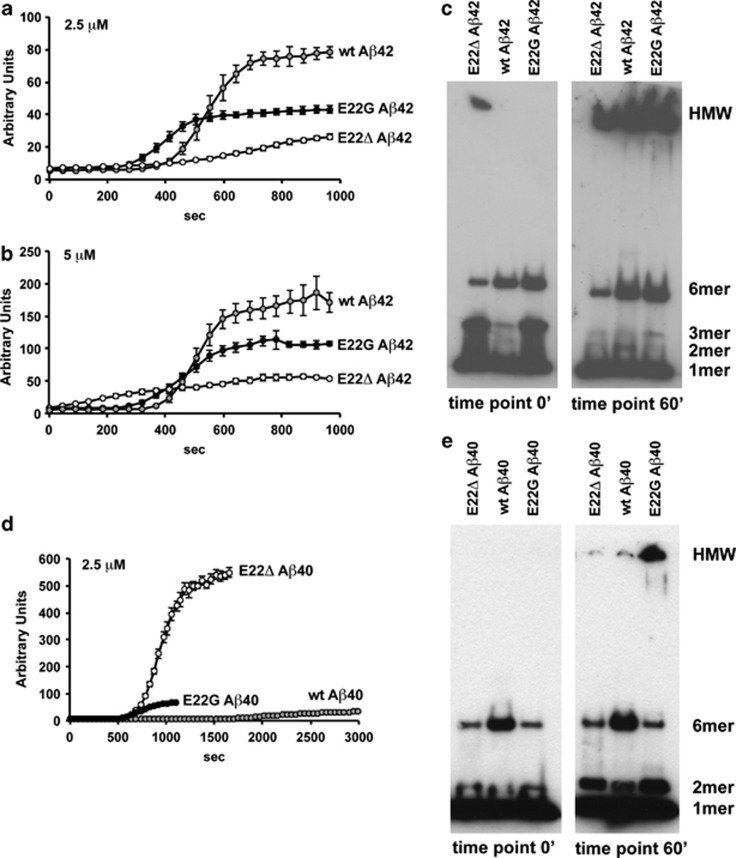
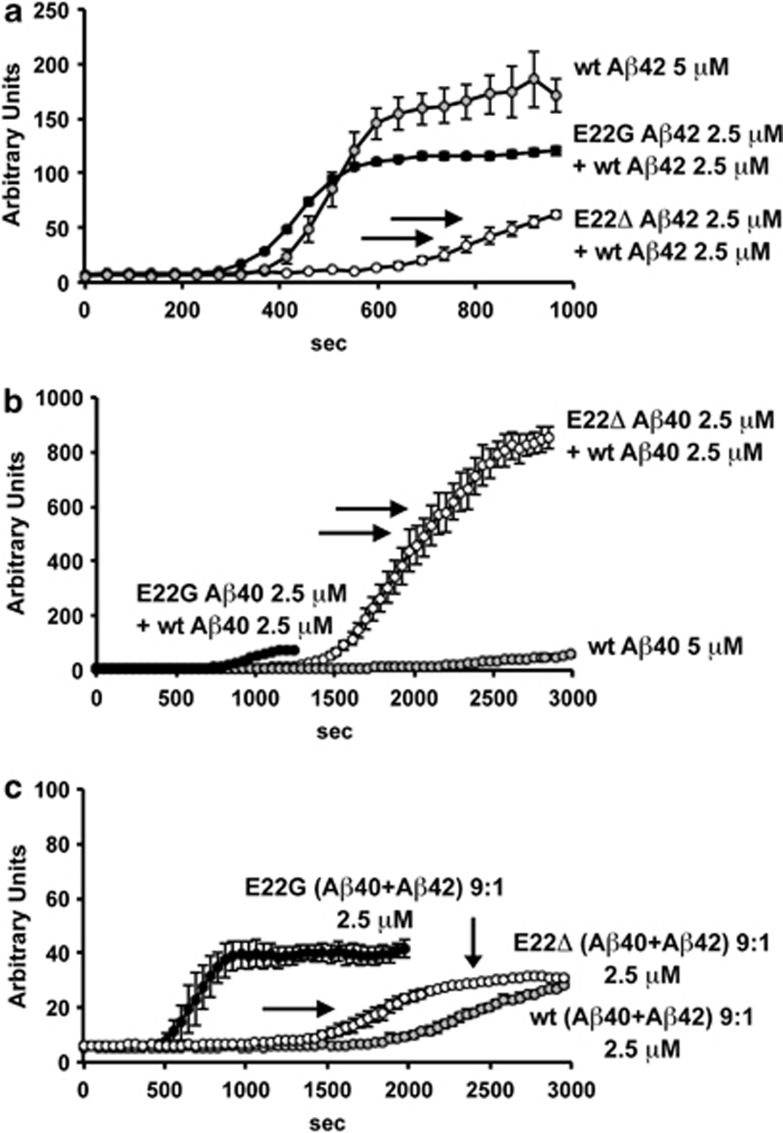
Pathogenic amyloid-β peptide precursor (APP) mutations clustered around position 693 of APP-position 22 of the Aβ sequence--are commonly associated with congophilic amyloid angiopathy (CAA) and intracerebral hemorrhages. In contrast, the Osaka (E693Δ) intra-Aβ APP mutation shows a recessive pattern of inheritance that leads to AD-like dementia despite low brain amyloid on in vivo positron emission tomography imaging. Here, we investigated the effects of the Osaka APP mutation on Aβ accumulation and deposition in vivo using a newly generated APP transgenic mouse model (E22ΔAβ) expressing the Osaka mutation together with the Swedish (K670N/M671L) double mutation. E22ΔAβ mice exhibited reduced α-processing of APP and early accumulation of intraneuronal fibrillar Aβ oligomers associated with cognitive deficits. In line with our in vitro findings that recombinant E22Δ-mutated Aβ peptides form amyloid fibrils, aged E22ΔAβ mice showed extracellular CAA deposits in leptomeningeal cerebellar and cortical vessels. In vitro results from thioflavin T aggregation assays with recombinant Aβ peptides revealed a yet unknown antiamyloidogenic property of the E693Δ mutation in the heterozygous state and an inhibitory effect of E22Δ Aβ42 on E22Δ Aβ40 fibrillogenesis. Moreover, E22Δ Aβ42 showed a unique aggregation kinetics lacking exponential fibril growth and poor seeding effects on wild-type Aβ aggregation. These results provide a possible explanation for the recessive trait of inheritance of the Osaka APP mutation and the apparent lack of amyloid deposition in E693Δ mutation carriers.
Figures
Similar articles
-
Familial Alzheimer's disease mutations at position 22 of the amyloid β-peptide sequence differentially affect synaptic loss, tau phosphorylation and neuronal cell death in an ex vivo system.PLoS One. 2020 Sep 23;15(9):e0239584. doi: 10.1371/journal.pone.0239584. eCollection 2020. PLoS One. 2020. PMID: 32966331 Free PMC article.
-
Alzheimer's disease amyloid β-protein mutations and deletions that define neuronal binding/internalization as early stage nonfibrillar/fibrillar aggregates and late stage fibrils.Biochemistry. 2012 May 15;51(19):3993-4003. doi: 10.1021/bi300275g. Epub 2012 May 7. Biochemistry. 2012. PMID: 22545812
-
Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models.Acta Neuropathol Commun. 2015 Nov 10;3:70. doi: 10.1186/s40478-015-0250-y. Acta Neuropathol Commun. 2015. PMID: 26556230 Free PMC article.
-
APP Osaka Mutation in Familial Alzheimer's Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance.Int J Mol Sci. 2020 Feb 19;21(4):1413. doi: 10.3390/ijms21041413. Int J Mol Sci. 2020. PMID: 32093100 Free PMC article. Review.
-
Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models.Int J Mol Sci. 2009 Apr 23;10(4):1872-1895. doi: 10.3390/ijms10041872. Int J Mol Sci. 2009. PMID: 19468344 Free PMC article. Review.
Cited by
-
Early onset Alzheimer's disease and oxidative stress.Oxid Med Cell Longev. 2014;2014:375968. doi: 10.1155/2014/375968. Epub 2014 Jan 14. Oxid Med Cell Longev. 2014. PMID: 24669286 Free PMC article. Review.
-
Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques.Neurobiol Dis. 2014 Nov;71:53-61. doi: 10.1016/j.nbd.2014.07.011. Epub 2014 Aug 1. Neurobiol Dis. 2014. PMID: 25092575 Free PMC article.
-
Distinct changes in all major components of the neurovascular unit across different neuropathological stages of Alzheimer's disease.Brain Pathol. 2020 Nov;30(6):1056-1070. doi: 10.1111/bpa.12895. Epub 2020 Sep 16. Brain Pathol. 2020. PMID: 32866303 Free PMC article.
-
Insulin-like growth factor-1 improves postoperative cognitive dysfunction following splenectomy in aged rats.Exp Ther Med. 2021 Mar;21(3):215. doi: 10.3892/etm.2021.9647. Epub 2021 Jan 15. Exp Ther Med. 2021. PMID: 33574912 Free PMC article.
-
Novel Phosphorylation-State Specific Antibodies Reveal Differential Deposition of Ser26 Phosphorylated Aβ Species in a Mouse Model of Alzheimer's Disease.Front Mol Neurosci. 2021 Jan 15;13:619639. doi: 10.3389/fnmol.2020.619639. eCollection 2020. Front Mol Neurosci. 2021. PMID: 33519377 Free PMC article.
References
-
- Thal DR, Del Tredici K, Braak H. Neurodegeneration in normal brain aging and disease. Sci Aging Knowledge Environ. 2004;2004:pe26. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. - PubMed
-
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
