

DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria
- PMID: 23141293
- PMCID: PMC3516599
- DOI: 10.1016/j.ajhg.2012.10.006
DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria
Abstract
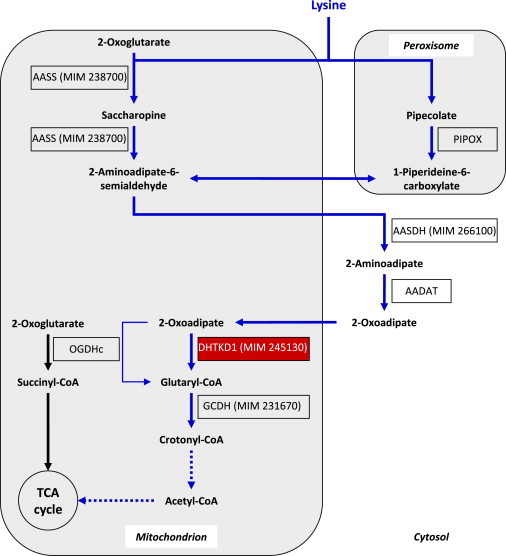
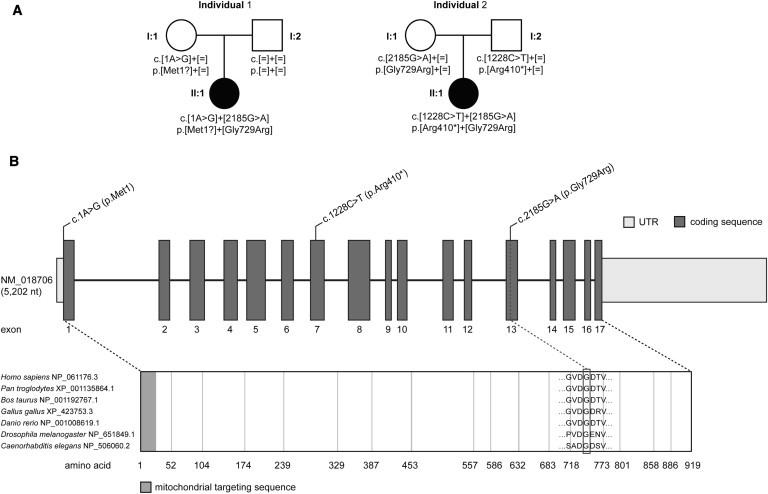
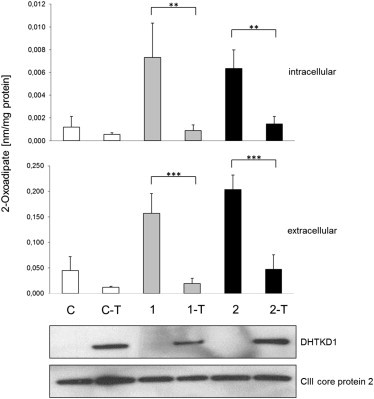
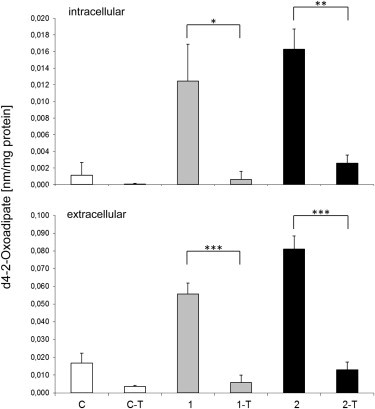
Abnormalities in metabolite profiles are valuable indicators of underlying pathologic conditions at the molecular level. However, their interpretation relies on detailed knowledge of the pathways, enzymes, and genes involved. Identification and characterization of their physiological function are therefore crucial for our understanding of human disease: they can provide guidance for therapeutic intervention and help us to identify suitable biomarkers for monitoring associated disorders. We studied two individuals with 2-aminoadipic and 2-oxoadipic aciduria, a metabolic condition that is still unresolved at the molecular level. This disorder has been associated with varying neurological symptoms. Exome sequencing of a single affected individual revealed compound heterozygosity for an initiating methionine mutation (c.1A>G) and a missense mutation (c.2185G>A [p.Gly729Arg]) in DHTKD1. This gene codes for dehydrogenase E1 and transketolase domain-containing protein 1, which is part of a 2-oxoglutarate-dehydrogenase-complex-like protein. Sequence analysis of a second individual identified the same missense mutation together with a nonsense mutation (c.1228C>T [p.Arg410(∗)]) in DHTKD1. Increased levels of 2-oxoadipate in individual-derived fibroblasts normalized upon lentiviral expression of the wild-type DHTKD1 mRNA. Moreover, investigation of L-lysine metabolism showed an accumulation of deuterium-labeled 2-oxoadipate only in noncomplemented cells, demonstrating that DHTKD1 codes for the enzyme mediating the last unresolved step in the L-lysine-degradation pathway. All together, our results establish mutations in DHTKD1 as a cause of human 2-aminoadipic and 2-oxoadipic aciduria via impaired turnover of decarboxylation 2-oxoadipate to glutaryl-CoA.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Human 2-Oxoglutarate Dehydrogenase and 2-Oxoadipate Dehydrogenase Both Generate Superoxide/H2O2 in a Side Reaction and Each Could Contribute to Oxidative Stress in Mitochondria.Neurochem Res. 2019 Oct;44(10):2325-2335. doi: 10.1007/s11064-019-02765-w. Epub 2019 Mar 7. Neurochem Res. 2019. PMID: 30847859 Review.
-
Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria.J Inherit Metab Dis. 2015 Sep;38(5):873-9. doi: 10.1007/s10545-015-9841-9. Epub 2015 Apr 10. J Inherit Metab Dis. 2015. PMID: 25860818
-
Elevated glutaric acid levels in Dhtkd1-/Gcdh- double knockout mice challenge our current understanding of lysine metabolism.Biochim Biophys Acta Mol Basis Dis. 2017 Sep;1863(9):2220-2228. doi: 10.1016/j.bbadis.2017.05.018. Epub 2017 May 22. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 28545977
-
DHTKD1 Deficiency Causes Charcot-Marie-Tooth Disease in Mice.Mol Cell Biol. 2018 Jun 14;38(13):e00085-18. doi: 10.1128/MCB.00085-18. Print 2018 Jul 1. Mol Cell Biol. 2018. PMID: 29661920 Free PMC article.
-
[Inborn errors of lysine metabolism].Ann Biol Clin (Paris). 1991;49(1):27-35. Ann Biol Clin (Paris). 1991. PMID: 1904694 Review. French.
Cited by
-
Heterozygous DHTKD1 Variants in Two European Cohorts of Amyotrophic Lateral Sclerosis Patients.Genes (Basel). 2021 Dec 29;13(1):84. doi: 10.3390/genes13010084. Genes (Basel). 2021. PMID: 35052424 Free PMC article.
-
Human 2-Oxoglutarate Dehydrogenase and 2-Oxoadipate Dehydrogenase Both Generate Superoxide/H2O2 in a Side Reaction and Each Could Contribute to Oxidative Stress in Mitochondria.Neurochem Res. 2019 Oct;44(10):2325-2335. doi: 10.1007/s11064-019-02765-w. Epub 2019 Mar 7. Neurochem Res. 2019. PMID: 30847859 Review.
-
Tissue metabolic profiling shows that saccharopine accumulates during renal ischemic-reperfusion injury, while kynurenine and itaconate accumulate in renal allograft rejection.Metabolomics. 2020 May 4;16(5):65. doi: 10.1007/s11306-020-01682-2. Metabolomics. 2020. PMID: 32367163 Free PMC article.
-
Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population.Cell. 2014 Sep 11;158(6):1415-1430. doi: 10.1016/j.cell.2014.07.039. Cell. 2014. PMID: 25215496 Free PMC article.
-
α-Ketoadipic Acid and α-Aminoadipic Acid Cause Disturbance of Glutamatergic Neurotransmission and Induction of Oxidative Stress In Vitro in Brain of Adolescent Rats.Neurotox Res. 2017 Aug;32(2):276-290. doi: 10.1007/s12640-017-9735-8. Epub 2017 Apr 20. Neurotox Res. 2017. PMID: 28429309
References
-
- Przyrembel H., Bachmann D., Lombeck I., Becker K., Wendel U., Wadman S.K., Bremer H.J. Alpha-ketoadipic aciduria, a new inborn error of lysine metabolism; biochemical studies. Clin. Chim. Acta. 1975;58:257–269. - PubMed
-
- Duran M., Beemer F.A., Wadman S.K., Wendel U., Janssen B. A patient with alpha-ketoadipic and alpha-aminoadipic aciduria. J. Inherit. Metab. Dis. 1984;7:61. - PubMed
-
- Sauer S.W., Opp S., Hoffmann G.F., Koeller D.M., Okun J.G., Kölker S. Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain. 2011;134:157–170. - PubMed
-
- Rustin P., Bourgeron T., Parfait B., Chretien D., Munnich A., Rötig A. Inborn errors of the Krebs cycle: A group of unusual mitochondrial diseases in human. Biochim. Biophys. Acta. 1997;1361:185–197. - PubMed
-
- Fiermonte G., Dolce V., Palmieri L., Ventura M., Runswick M.J., Palmieri F., Walker J.E. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001;276:8225–8230. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
