

Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice
- PMID: 23038755
- PMCID: PMC3528309
- DOI: 10.1096/fj.12-208660
Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice
Abstract
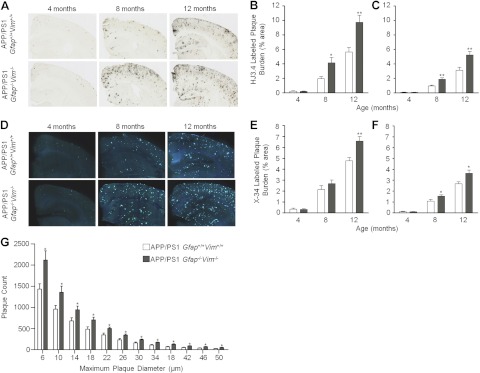
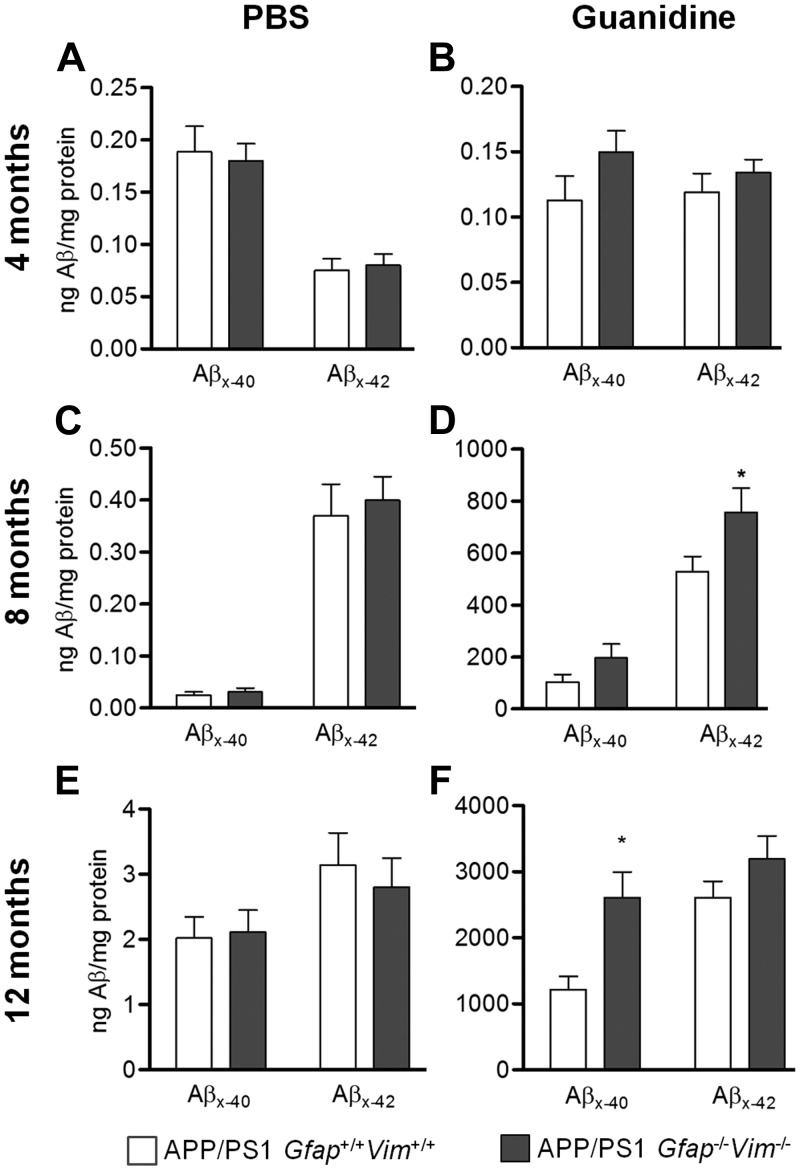
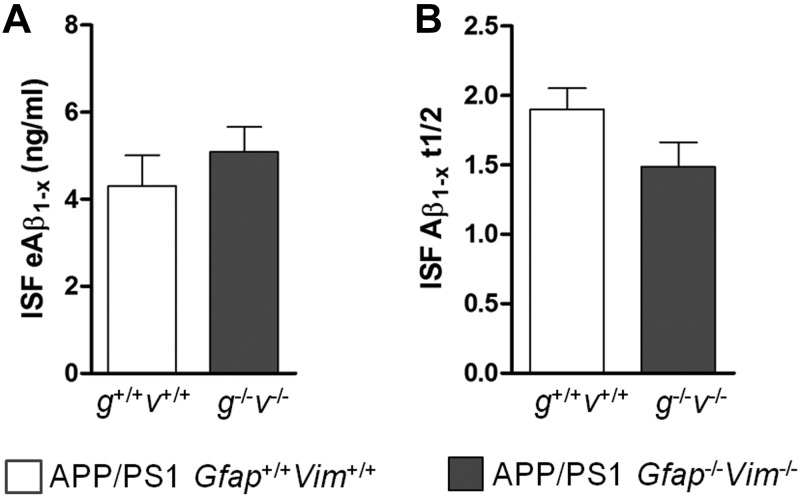
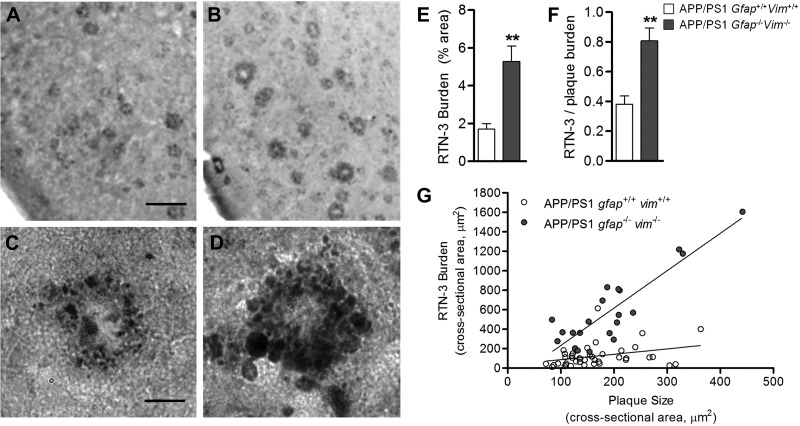
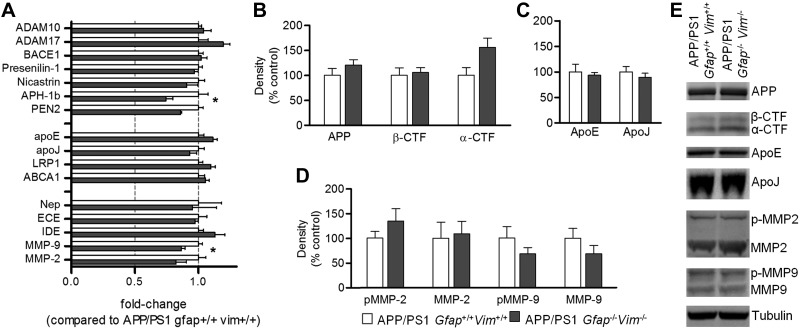
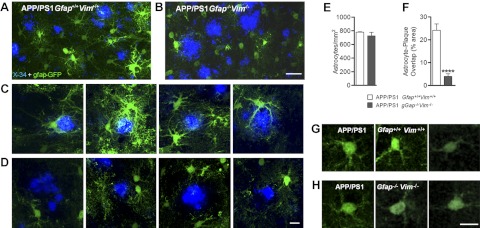
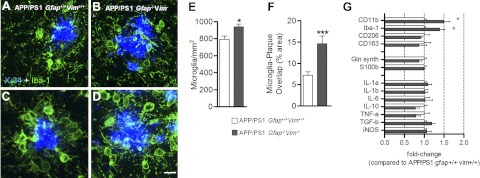
The accumulation of aggregated amyloid-β (Aβ) in amyloid plaques is a neuropathological hallmark of Alzheimer's disease (AD). Reactive astrocytes are intimately associated with amyloid plaques; however, their role in AD pathogenesis is unclear. We deleted the genes encoding two intermediate filament proteins required for astrocyte activation-glial fibrillary acid protein (Gfap) and vimentin (Vim)-in transgenic mice expressing mutant human amyloid precursor protein and presenilin-1 (APP/PS1). The gene deletions increased amyloid plaque load: APP/PS1 Gfap(-/-)Vim(-/-) mice had twice the plaque load of APP/PS1 Gfap(+/+)Vim(+/+) mice at 8 and 12 mo of age. APP expression and soluble and interstitial fluid Aβ levels were unchanged, suggesting that the deletions had no effect on APP processing or Aβ generation. Astrocyte morphology was markedly altered by the deletions: wild-type astrocytes had hypertrophied processes that surrounded and infiltrated plaques, whereas Gfap(-/-)Vim(-/-) astrocytes had little process hypertrophy and lacked contact with adjacent plaques. Moreover, Gfap and Vim gene deletion resulted in a marked increase in dystrophic neurites (2- to 3-fold higher than APP/PS1 Gfap(+/+)Vim(+/+) mice), even after normalization for amyloid load. These results suggest that astrocyte activation limits plaque growth and attenuates plaque-related dystrophic neurites. These activities may require intimate contact between astrocyte and plaque.
Figures
Similar articles
-
GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer's disease.Glia. 2015 Jun;63(6):1036-56. doi: 10.1002/glia.22800. Epub 2015 Mar 2. Glia. 2015. PMID: 25731615
-
Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis.J Neurosci. 2014 Jul 16;34(29):9607-20. doi: 10.1523/JNEUROSCI.3788-13.2014. J Neurosci. 2014. PMID: 25031402 Free PMC article.
-
Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer's disease mouse model with suppression of glymphatic clearance.Alzheimers Res Ther. 2020 Oct 2;12(1):125. doi: 10.1186/s13195-020-00688-1. Alzheimers Res Ther. 2020. PMID: 33008458 Free PMC article.
-
Characterization of astrocytes throughout life in wildtype and APP/PS1 mice after early-life stress exposure.J Neuroinflammation. 2020 Mar 20;17(1):91. doi: 10.1186/s12974-020-01762-z. J Neuroinflammation. 2020. PMID: 32197653 Free PMC article.
-
Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice.Alzheimers Res Ther. 2021 Feb 9;13(1):40. doi: 10.1186/s13195-020-00761-9. Alzheimers Res Ther. 2021. PMID: 33563332 Free PMC article.
Cited by
-
Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer's Disease?CNS Neurosci Ther. 2019 Jan;25(1):5-13. doi: 10.1111/cns.13050. Epub 2018 Aug 12. CNS Neurosci Ther. 2019. PMID: 30101571 Free PMC article. Review.
-
Astrocyte Activation and the Calcineurin/NFAT Pathway in Cerebrovascular Disease.Front Aging Neurosci. 2018 Sep 21;10:287. doi: 10.3389/fnagi.2018.00287. eCollection 2018. Front Aging Neurosci. 2018. PMID: 30297999 Free PMC article. Review.
-
Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke.Glia. 2014 Dec;62(12):2022-33. doi: 10.1002/glia.22723. Epub 2014 Jul 15. Glia. 2014. PMID: 25043249 Free PMC article.
-
Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease.Neuropharmacology. 2015 Sep;96(Pt A):29-41. doi: 10.1016/j.neuropharm.2014.10.028. Epub 2014 Nov 13. Neuropharmacology. 2015. PMID: 25445485 Free PMC article. Review.
-
TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide.Mol Neurodegener. 2017 Jul 6;12(1):53. doi: 10.1186/s13024-017-0194-8. Mol Neurodegener. 2017. PMID: 28683776 Free PMC article.
References
-
- Weisman D., Hakimian E., Ho G. J. (2006) Interleukins, inflammation, and mechanisms of Alzheimer's disease. Vitam. Horm. 74, 505–530 - PubMed
-
- Wyss-Coray T. (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015 - PubMed
-
- DiCarlo G., Wilcock D., Henderson D., Gordon M., Morgan D. (2001) Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol. Aging 22, 1007–1012 - PubMed
-
- Chakrabarty P., Jansen-West K., Beccard A., Ceballos-Diaz C., Levites Y., Verbeeck C., Zubair A. C., Dickson D., Golde T. E., Das P. (2009) Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24, 548–559 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
