

Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease
- PMID: 23028132
- PMCID: PMC3475405
- DOI: 10.1101/cshperspect.a011452
Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease
Abstract
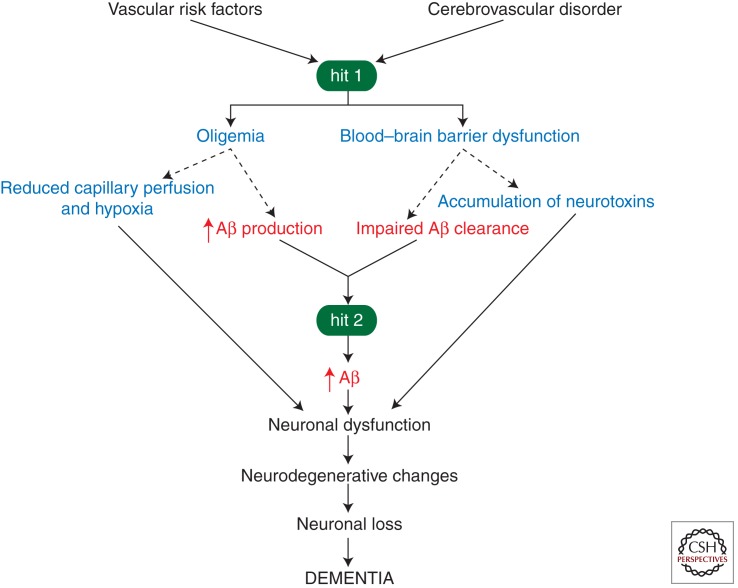
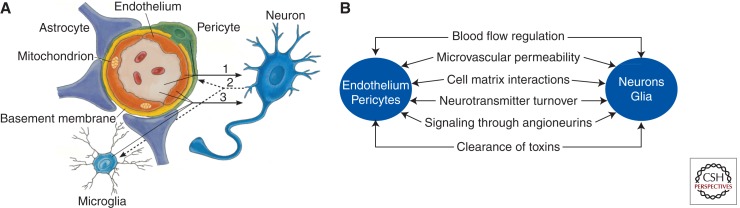
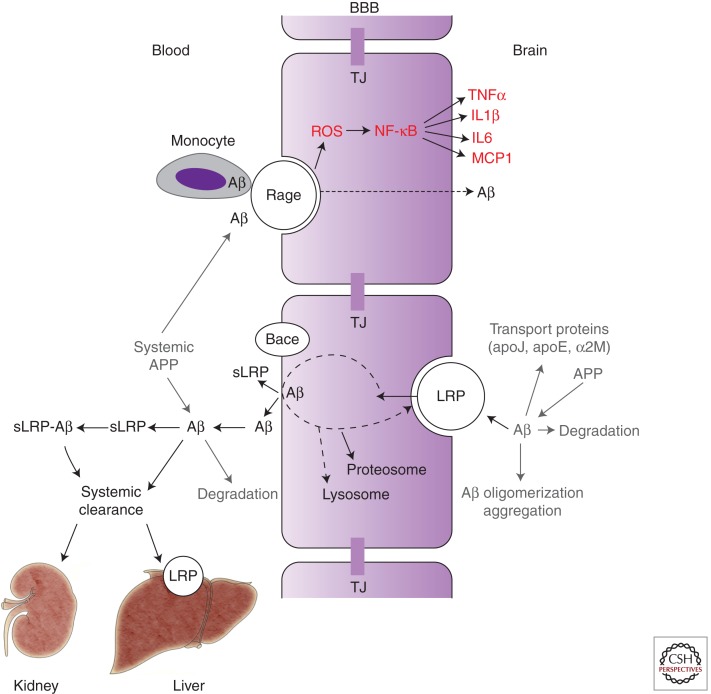
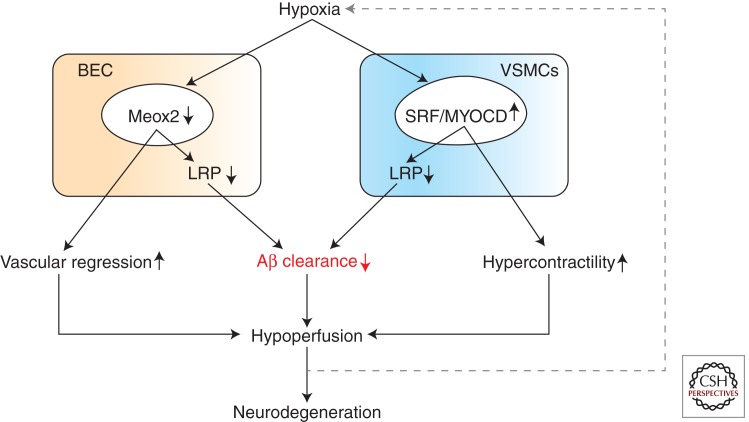
Neurovascular dysfunction is an integral part of Alzheimer disease (AD). Changes in the brain vascular system may contribute in a significant way to the onset and progression of cognitive decline and the development of a chronic neurodegenerative process associated with accumulation of amyloid β-peptide (Aβ) in brain and cerebral vessels in AD individuals and AD animal models. Here, we review the role of the neurovascular unit and molecular mechanisms in cerebral vascular cells behind the pathogenesis of AD. In particular, we focus on blood-brain barrier (BBB) dysfunction, decreased cerebral blood flow, and impaired vascular clearance of Aβ from brain. The data reviewed here support an essential role of the neurovascular and BBB mechanisms in AD pathogenesis.
Figures
Similar articles
-
Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease.Acta Neuropathol. 2009 Jul;118(1):103-13. doi: 10.1007/s00401-009-0522-3. Epub 2009 Mar 25. Acta Neuropathol. 2009. PMID: 19319544 Free PMC article. Review.
-
Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease.J Alzheimers Dis. 2013;33 Suppl 1(0 1):S87-100. doi: 10.3233/JAD-2012-129037. J Alzheimers Dis. 2013. PMID: 22751174 Free PMC article. Review.
-
Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease.Curr Alzheimer Res. 2007 Apr;4(2):191-7. doi: 10.2174/156720507780362245. Curr Alzheimer Res. 2007. PMID: 17430246 Review.
-
New therapeutic targets in the neurovascular pathway in Alzheimer's disease.Neurotherapeutics. 2008 Jul;5(3):409-14. doi: 10.1016/j.nurt.2008.05.011. Neurotherapeutics. 2008. PMID: 18625452 Free PMC article. Review.
-
Neurovascular mechanisms of Alzheimer's neurodegeneration.Trends Neurosci. 2005 Apr;28(4):202-8. doi: 10.1016/j.tins.2005.02.001. Trends Neurosci. 2005. PMID: 15808355 Review.
Cited by
-
Physiological and Pathological Remodeling of Cerebral Microvessels.Int J Mol Sci. 2022 Oct 21;23(20):12683. doi: 10.3390/ijms232012683. Int J Mol Sci. 2022. PMID: 36293539 Free PMC article. Review.
-
Linking Cerebrovascular Dysfunction to Age-Related Hearing Loss and Alzheimer's Disease-Are Systemic Approaches for Diagnosis and Therapy Required?Biomolecules. 2022 Nov 19;12(11):1717. doi: 10.3390/biom12111717. Biomolecules. 2022. PMID: 36421731 Free PMC article. Review.
-
The pericyte: A critical cell in the pathogenesis of CADASIL.Cereb Circ Cogn Behav. 2021;2:100031. doi: 10.1016/j.cccb.2021.100031. Cereb Circ Cogn Behav. 2021. PMID: 34950895 Free PMC article. Review.
-
Blood-brain-barriers in aging and in Alzheimer's disease.Mol Neurodegener. 2013 Oct 22;8:38. doi: 10.1186/1750-1326-8-38. Mol Neurodegener. 2013. PMID: 24148264 Free PMC article. Review.
-
Arterial Stiffness Due to Carotid Calcification Disrupts Cerebral Blood Flow Regulation and Leads to Cognitive Deficits.J Am Heart Assoc. 2019 May 7;8(9):e011630. doi: 10.1161/JAHA.118.011630. J Am Heart Assoc. 2019. PMID: 31057061 Free PMC article.
References
-
- Arelin K, Kinoshita A, Whelan CM, Irizarry MC, Rebeck GW, Strickland DK, Hyman BT 2002. LRP and senile plaques in Alzheimer’s disease: Colocalization with apolipoprotein E and with activated astrocytes. Brain Res Mol Brain Res 104: 38–46 - PubMed
-
- Atwood CS, Bishop GM, Perry G, Smith MA 2002. Amyloid-β: A vascular sealant that protects against hemorrhage? J Neurosci Res 70: 356. - PubMed
-
- Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV 2002. Brain clearance of Alzheimer’s amyloid-β40 in the squirrel monkey: A SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target 10: 359–368 - PubMed
-
- Bailey TL, Rivara CB, Rocher AB, Hof PR 2004. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 26: 573–578 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical