

Statistical inference of the generation probability of T-cell receptors from sequence repertoires
- PMID: 22988065
- PMCID: PMC3479580
- DOI: 10.1073/pnas.1212755109
Statistical inference of the generation probability of T-cell receptors from sequence repertoires
Abstract
Stochastic rearrangement of germline V-, D-, and J-genes to create variable coding sequence for certain cell surface receptors is at the origin of immune system diversity. This process, known as "VDJ recombination", is implemented via a series of stochastic molecular events involving gene choices and random nucleotide insertions between, and deletions from, genes. We use large sequence repertoires of the variable CDR3 region of human CD4+ T-cell receptor beta chains to infer the statistical properties of these basic biochemical events. Because any given CDR3 sequence can be produced in multiple ways, the probability distribution of hidden recombination events cannot be inferred directly from the observed sequences; we therefore develop a maximum likelihood inference method to achieve this end. To separate the properties of the molecular rearrangement mechanism from the effects of selection, we focus on nonproductive CDR3 sequences in T-cell DNA. We infer the joint distribution of the various generative events that occur when a new T-cell receptor gene is created. We find a rich picture of correlation (and absence thereof), providing insight into the molecular mechanisms involved. The generative event statistics are consistent between individuals, suggesting a universal biochemical process. Our probabilistic model predicts the generation probability of any specific CDR3 sequence by the primitive recombination process, allowing us to quantify the potential diversity of the T-cell repertoire and to understand why some sequences are shared between individuals. We argue that the use of formal statistical inference methods, of the kind presented in this paper, will be essential for quantitative understanding of the generation and evolution of diversity in the adaptive immune system.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
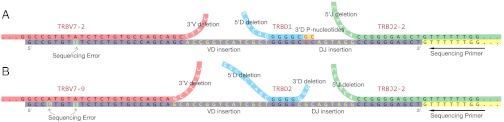
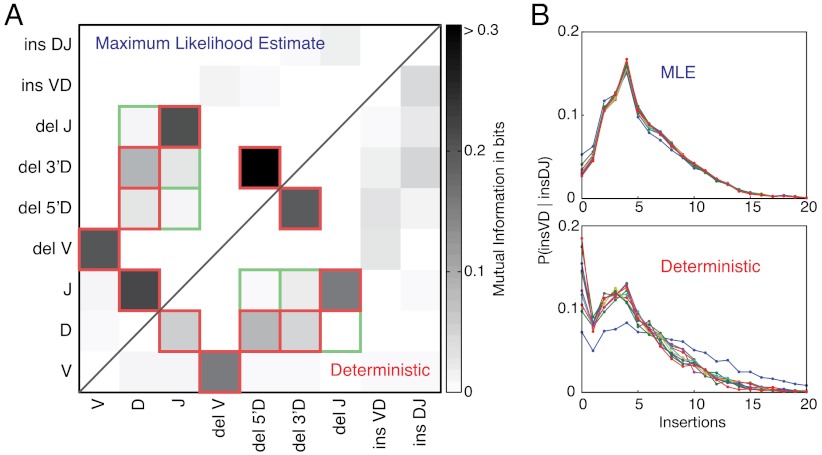
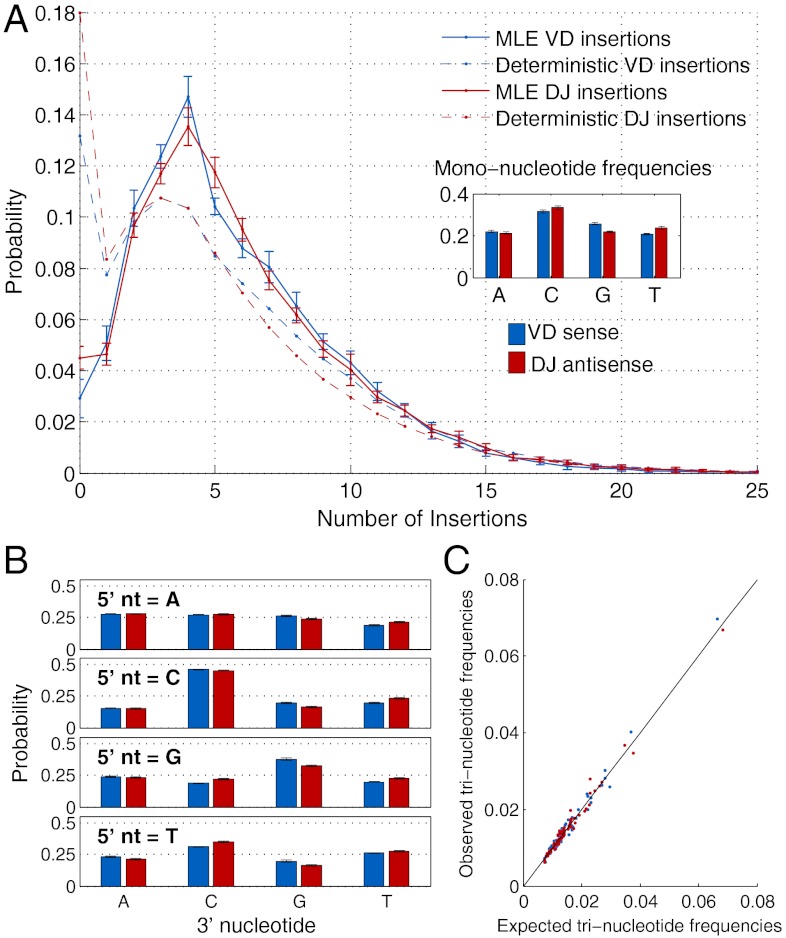
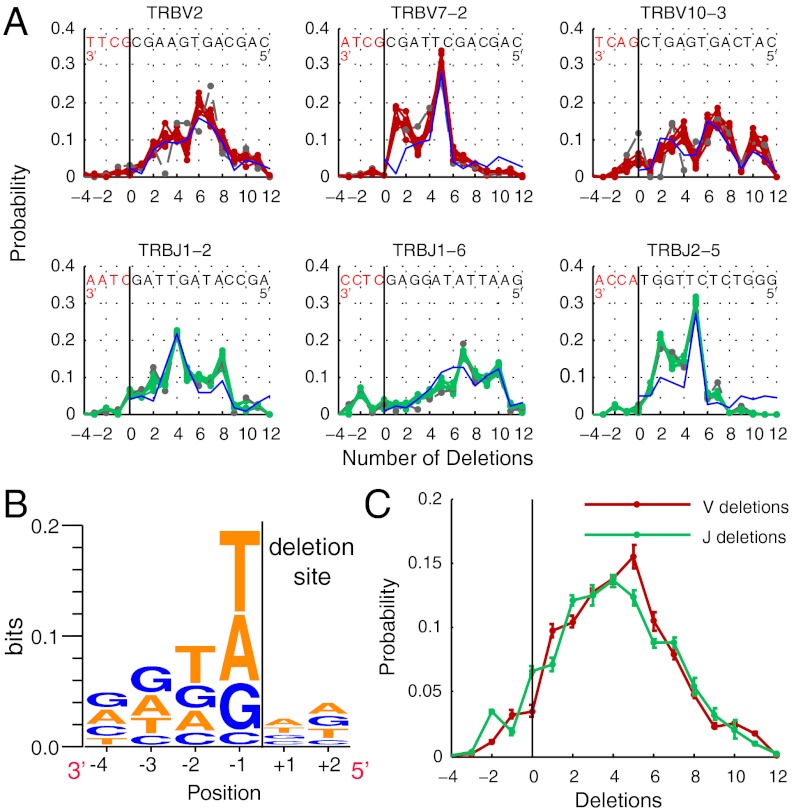
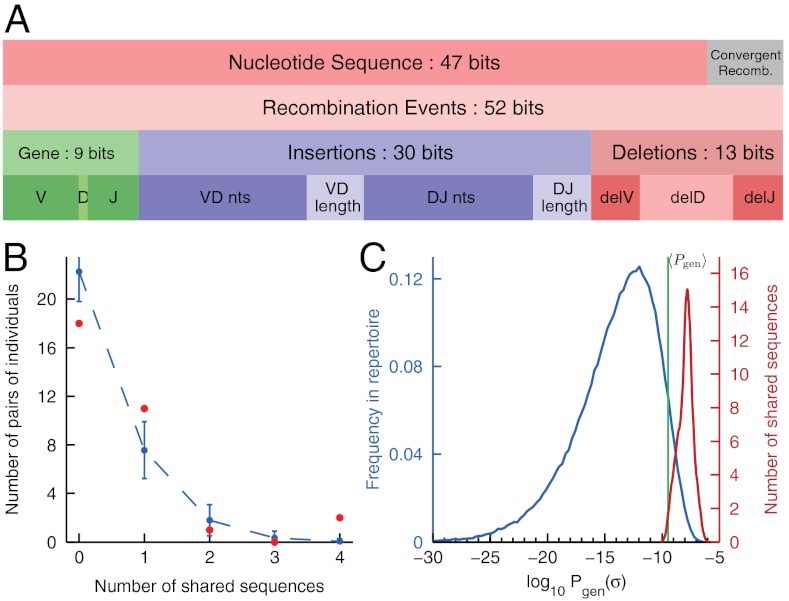
 for the full repertoire is indicated by the vertical green line.
for the full repertoire is indicated by the vertical green line.Similar articles
-
Inferring processes underlying B-cell repertoire diversity.Philos Trans R Soc Lond B Biol Sci. 2015 Sep 5;370(1676):20140243. doi: 10.1098/rstb.2014.0243. Philos Trans R Soc Lond B Biol Sci. 2015. PMID: 26194757 Free PMC article.
-
Insights into immune system development and function from mouse T-cell repertoires.Proc Natl Acad Sci U S A. 2017 Feb 28;114(9):2253-2258. doi: 10.1073/pnas.1700241114. Epub 2017 Feb 14. Proc Natl Acad Sci U S A. 2017. PMID: 28196891 Free PMC article.
-
repgenHMM: a dynamic programming tool to infer the rules of immune receptor generation from sequence data.Bioinformatics. 2016 Jul 1;32(13):1943-51. doi: 10.1093/bioinformatics/btw112. Epub 2016 Feb 26. Bioinformatics. 2016. PMID: 27153709 Free PMC article.
-
Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination.Immunol Rev. 2018 Jul;284(1):167-179. doi: 10.1111/imr.12665. Immunol Rev. 2018. PMID: 29944757 Free PMC article. Review.
-
The Bayesian optimist's guide to adaptive immune receptor repertoire analysis.Immunol Rev. 2018 Jul;284(1):148-166. doi: 10.1111/imr.12664. Immunol Rev. 2018. PMID: 29944760 Free PMC article. Review.
Cited by
-
The astonishing diversity of Ig classes and B cell repertoires in teleost fish.Front Immunol. 2013 Feb 13;4:28. doi: 10.3389/fimmu.2013.00028. eCollection 2013. Front Immunol. 2013. PMID: 23408183 Free PMC article.
-
Deep generative selection models of T and B cell receptor repertoires with soNNia.Proc Natl Acad Sci U S A. 2021 Apr 6;118(14):e2023141118. doi: 10.1073/pnas.2023141118. Proc Natl Acad Sci U S A. 2021. PMID: 33795515 Free PMC article.
-
Learning from HIV-1 to predict the immunogenicity of T cell epitopes in SARS-CoV-2.iScience. 2021 Apr 23;24(4):102311. doi: 10.1016/j.isci.2021.102311. Epub 2021 Mar 15. iScience. 2021. PMID: 33748696 Free PMC article.
-
Preselection TCR repertoire predicts CD4+ and CD8+ T-cell differentiation state.Immunology. 2020 Dec;161(4):354-363. doi: 10.1111/imm.13256. Epub 2020 Oct 7. Immunology. 2020. PMID: 32875554 Free PMC article.
-
Urine-derived lymphocytes as a non-invasive measure of the bladder tumor immune microenvironment.J Exp Med. 2018 Nov 5;215(11):2748-2759. doi: 10.1084/jem.20181003. Epub 2018 Sep 26. J Exp Med. 2018. PMID: 30257862 Free PMC article.
References
-
- Murphy KP, Travers P, Walport M, Janeway C. Janeway’s Immunobiology. New York: Garland; 2008.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
