

Genome-wide association between DNA methylation and alternative splicing in an invertebrate
- PMID: 22978521
- PMCID: PMC3526459
- DOI: 10.1186/1471-2164-13-480
Genome-wide association between DNA methylation and alternative splicing in an invertebrate
Abstract
Background: Gene bodies are the most evolutionarily conserved targets of DNA methylation in eukaryotes. However, the regulatory functions of gene body DNA methylation remain largely unknown. DNA methylation in insects appears to be primarily confined to exons. Two recent studies in Apis mellifera (honeybee) and Nasonia vitripennis (jewel wasp) analyzed transcription and DNA methylation data for one gene in each species to demonstrate that exon-specific DNA methylation may be associated with alternative splicing events. In this study we investigated the relationship between DNA methylation, alternative splicing, and cross-species gene conservation on a genome-wide scale using genome-wide transcription and DNA methylation data.
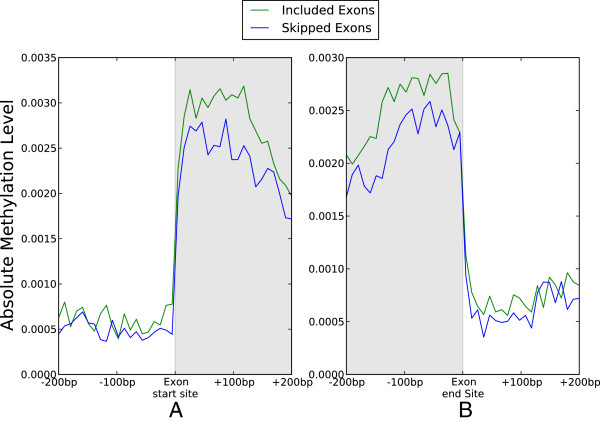
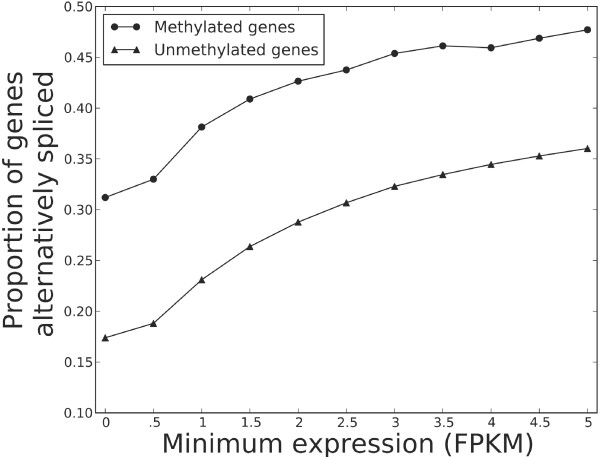
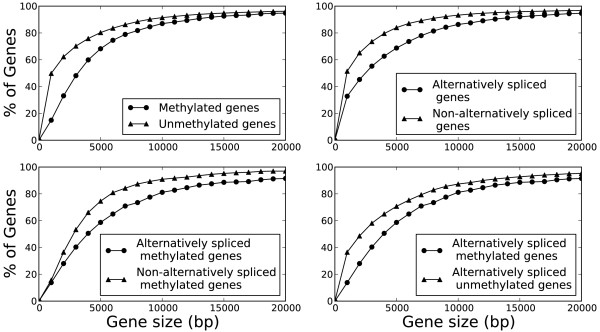
Results: We generated RNA deep sequencing data (RNA-seq) to measure genome-wide mRNA expression at the exon- and gene-level. We produced a de novo transcriptome from this RNA-seq data and computationally predicted splice variants for the honeybee genome. We found that exons that are included in transcription are higher methylated than exons that are skipped during transcription. We detected enrichment for alternative splicing among methylated genes compared to unmethylated genes using fisher's exact test. We performed a statistical analysis to reveal that the presence of DNA methylation or alternative splicing are both factors associated with a longer gene length and a greater number of exons in genes. In concordance with this observation, a conservation analysis using BLAST revealed that each of these factors is also associated with higher cross-species gene conservation.
Conclusions: This study constitutes the first genome-wide analysis exhibiting a positive relationship between exon-level DNA methylation and mRNA expression in the honeybee. Our finding that methylated genes are enriched for alternative splicing suggests that, in invertebrates, exon-level DNA methylation may play a role in the construction of splice variants by positively influencing exon inclusion during transcription. The results from our cross-species homology analysis suggest that DNA methylation and alternative splicing are genetic mechanisms whose utilization could contribute to a longer gene length and a slower rate of gene evolution.
Figures
Similar articles
-
Function and evolution of DNA methylation in Nasonia vitripennis.PLoS Genet. 2013;9(10):e1003872. doi: 10.1371/journal.pgen.1003872. Epub 2013 Oct 10. PLoS Genet. 2013. PMID: 24130511 Free PMC article.
-
Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes.Mol Biol Evol. 2011 Dec;28(12):3345-54. doi: 10.1093/molbev/msr168. Epub 2011 Jun 20. Mol Biol Evol. 2011. PMID: 21693438 Free PMC article.
-
OGS2: genome re-annotation of the jewel wasp Nasonia vitripennis.BMC Genomics. 2016 Aug 25;17(1):678. doi: 10.1186/s12864-016-2886-9. BMC Genomics. 2016. PMID: 27561358 Free PMC article.
-
DNA Methylation and Gene Regulation in Honeybees: From Genome-Wide Analyses to Obligatory Epialleles.Adv Exp Med Biol. 2016;945:193-211. doi: 10.1007/978-3-319-43624-1_9. Adv Exp Med Biol. 2016. PMID: 27826840 Review.
-
The alternative role of DNA methylation in splicing regulation.Trends Genet. 2015 May;31(5):274-80. doi: 10.1016/j.tig.2015.03.002. Epub 2015 Mar 30. Trends Genet. 2015. PMID: 25837375 Review.
Cited by
-
A diverse epigenetic landscape at human exons with implication for expression.Nucleic Acids Res. 2015 Apr 20;43(7):3498-508. doi: 10.1093/nar/gkv153. Epub 2015 Mar 12. Nucleic Acids Res. 2015. PMID: 25765649 Free PMC article.
-
A Broad Survey of Gene Body and Repeat Methylation in Cnidaria Reveals a Complex Evolutionary History.Genome Biol Evol. 2022 Feb 4;14(2):evab284. doi: 10.1093/gbe/evab284. Genome Biol Evol. 2022. PMID: 35104341 Free PMC article.
-
Epigenetic studies in Alzheimer's disease: current findings, caveats, and considerations for future studies.Am J Med Genet B Neuropsychiatr Genet. 2013 Dec;162B(8):789-99. doi: 10.1002/ajmg.b.32201. Epub 2013 Sep 13. Am J Med Genet B Neuropsychiatr Genet. 2013. PMID: 24038819 Free PMC article. Review.
-
Gene Body Methylation Patterns in Daphnia Are Associated with Gene Family Size.Genome Biol Evol. 2016 Apr 25;8(4):1185-96. doi: 10.1093/gbe/evw069. Genome Biol Evol. 2016. PMID: 27017526 Free PMC article.
-
Abnormal DNA methylation may contribute to the progression of osteosarcoma.Mol Med Rep. 2018 Jan;17(1):193-199. doi: 10.3892/mmr.2017.7869. Epub 2017 Oct 25. Mol Med Rep. 2018. PMID: 29115427 Free PMC article.
References
-
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. - DOI - PMC - PubMed
-
- Xiang H, Zhu J, Chen Q, Dai F, Li X, Li M, Zhang H, Zhang G, Li D, Dong Y, Zhao L, Lin Y, Cheng D, Yu J, Sun J, Zhou X, Ma K, He Y, Zhao Y, Guo S, Ye M, Guo G, Li Y, Li R, Zhang X, Ma L, Kristiansen K, Guo Q, Jiang J, Beck S, Xia Q, Wang W, Wang J. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28:516–520. doi: 10.1038/nbt.1626. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
