

An integrated encyclopedia of DNA elements in the human genome
- PMID: 22955616
- PMCID: PMC3439153
- DOI: 10.1038/nature11247
An integrated encyclopedia of DNA elements in the human genome
Abstract
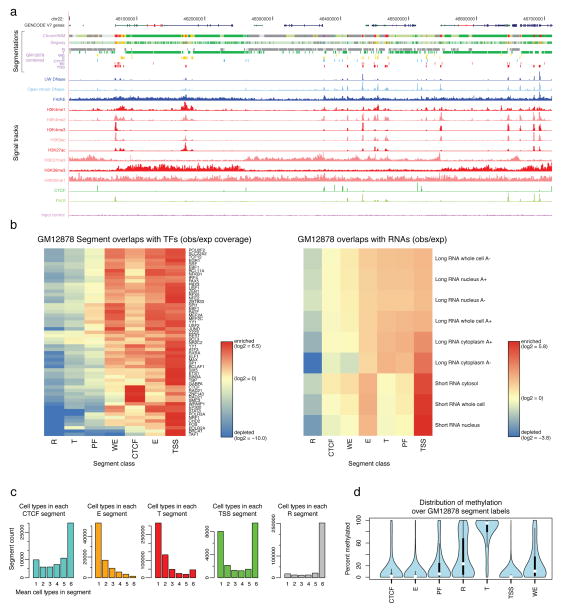
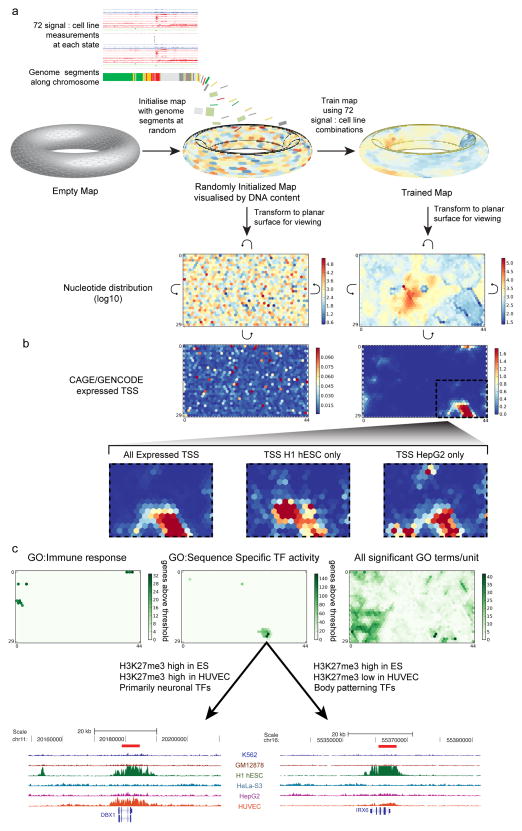
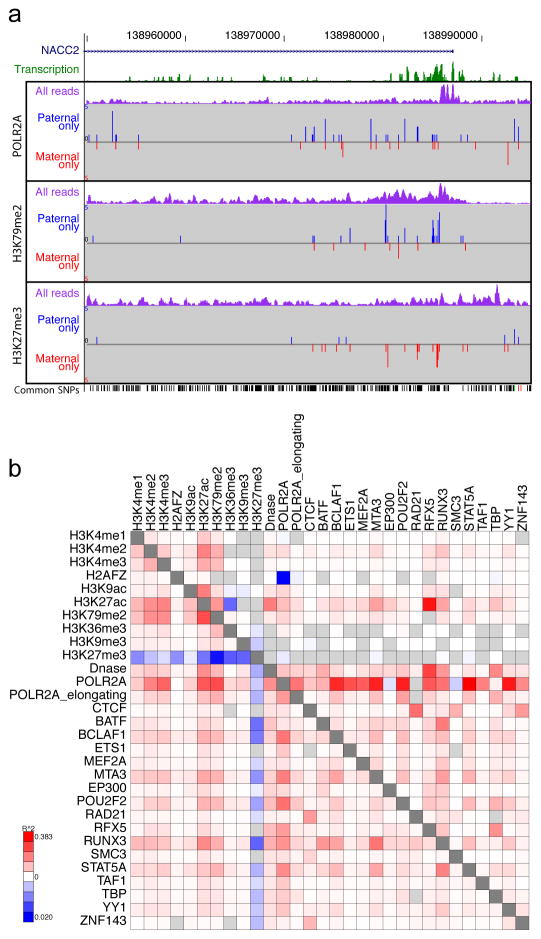
The human genome encodes the blueprint of life, but the function of the vast majority of its nearly three billion bases is unknown. The Encyclopedia of DNA Elements (ENCODE) project has systematically mapped regions of transcription, transcription factor association, chromatin structure and histone modification. These data enabled us to assign biochemical functions for 80% of the genome, in particular outside of the well-studied protein-coding regions. Many discovered candidate regulatory elements are physically associated with one another and with expressed genes, providing new insights into the mechanisms of gene regulation. The newly identified elements also show a statistical correspondence to sequence variants linked to human disease, and can thereby guide interpretation of this variation. Overall, the project provides new insights into the organization and regulation of our genes and genome, and is an expansive resource of functional annotations for biomedical research.
Figures
Comment in
-
Genomics: users' guide to the human genome.Nat Rev Genet. 2012 Oct;13(10):678. doi: 10.1038/nrg3329. Epub 2012 Sep 7. Nat Rev Genet. 2012. PMID: 22955793 No abstract available.
-
The ENCODE project.Nat Methods. 2012 Nov;9(11):1046. doi: 10.1038/nmeth.2238. Nat Methods. 2012. PMID: 23281567 No abstract available.
-
Literature watch: implications for transplantation.Am J Transplant. 2013 Feb;13(2):245. doi: 10.1111/ajt.12171. Am J Transplant. 2013. PMID: 23356894 No abstract available.
Similar articles
-
Cracking the ENCODE: from transcription to therapeutics.Hepatology. 2013 Jun;57(6):2532-5. doi: 10.1002/hep.26449. Hepatology. 2013. PMID: 23609523
-
The accessible chromatin landscape of the human genome.Nature. 2012 Sep 6;489(7414):75-82. doi: 10.1038/nature11232. Nature. 2012. PMID: 22955617 Free PMC article.
-
An expansive human regulatory lexicon encoded in transcription factor footprints.Nature. 2012 Sep 6;489(7414):83-90. doi: 10.1038/nature11212. Nature. 2012. PMID: 22955618 Free PMC article.
-
A brief review on the Human Encyclopedia of DNA Elements (ENCODE) project.Genomics Proteomics Bioinformatics. 2013 Jun;11(3):135-41. doi: 10.1016/j.gpb.2013.05.001. Epub 2013 May 28. Genomics Proteomics Bioinformatics. 2013. PMID: 23722115 Free PMC article. Review.
-
Epigenetics, chromatin and genome organization: recent advances from the ENCODE project.J Intern Med. 2014 Sep;276(3):201-14. doi: 10.1111/joim.12231. Epub 2014 Mar 27. J Intern Med. 2014. PMID: 24605849 Review.
Cited by
-
On the identification of differentially-active transcription factors from ATAC-seq data.PLoS Comput Biol. 2024 Oct 23;20(10):e1011971. doi: 10.1371/journal.pcbi.1011971. eCollection 2024 Oct. PLoS Comput Biol. 2024. PMID: 39441876 Free PMC article.
-
An ImmunoChip study of multiple sclerosis risk in African Americans.Brain. 2015 Jun;138(Pt 6):1518-30. doi: 10.1093/brain/awv078. Epub 2015 Mar 28. Brain. 2015. PMID: 25818868 Free PMC article.
-
A pooling-based approach to mapping genetic variants associated with DNA methylation.Genome Res. 2015 Jun;25(6):907-17. doi: 10.1101/gr.183749.114. Epub 2015 Apr 24. Genome Res. 2015. PMID: 25910490 Free PMC article.
-
Decoding ENCODE.Nat Chem Biol. 2012 Nov;8(11):871. doi: 10.1038/nchembio.1107. Nat Chem Biol. 2012. PMID: 23076055 No abstract available.
-
Crosstalk between epitranscriptomic and epigenomic modifications and its implication in human diseases.Cell Genom. 2024 Aug 14;4(8):100605. doi: 10.1016/j.xgen.2024.100605. Epub 2024 Jul 8. Cell Genom. 2024. PMID: 38981476 Free PMC article.
References
-
- Chiaromonte F, et al. The share of human genomic DNA under selection estimated from human-mouse genomic alignments. Cold Spring Harbor symposia on quantitative biology. 2003;68:245–254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01HG003143/HG/NHGRI NIH HHS/United States
- RC2 HG005573/HG/NHGRI NIH HHS/United States
- U54HG004592/HG/NHGRI NIH HHS/United States
- RC2 HG005679/HG/NHGRI NIH HHS/United States
- R01 HG003143/HG/NHGRI NIH HHS/United States
- ZIA HG200341/ImNIH/Intramural NIH HHS/United States
- U41HG004568/HG/NHGRI NIH HHS/United States
- U54 HG004557/HG/NHGRI NIH HHS/United States
- U01HG004695/HG/NHGRI NIH HHS/United States
- U01HG004561/HG/NHGRI NIH HHS/United States
- U54 HG004570/HG/NHGRI NIH HHS/United States
- R37 DK044746/DK/NIDDK NIH HHS/United States
- U54 HG004576/HG/NHGRI NIH HHS/United States
- R01HG003988/HG/NHGRI NIH HHS/United States
- U54 HG004592/HG/NHGRI NIH HHS/United States
- U54HG004576/HG/NHGRI NIH HHS/United States
- R01 HG003988/HG/NHGRI NIH HHS/United States
- R01 HG003541/HG/NHGRI NIH HHS/United States
- R01HG004456-03/HG/NHGRI NIH HHS/United States
- U54HG004558/HG/NHGRI NIH HHS/United States
- R01HG003541/HG/NHGRI NIH HHS/United States
- RC2HG005591/HG/NHGRI NIH HHS/United States
- RC2 HG005591/HG/NHGRI NIH HHS/United States
- K99 HG006698/HG/NHGRI NIH HHS/United States
- U54 HG004558/HG/NHGRI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- R01 DK054369/DK/NIDDK NIH HHS/United States
- ZIA HG200323/ImNIH/Intramural NIH HHS/United States
- U54HG004557/HG/NHGRI NIH HHS/United States
- U54 HG004563/HG/NHGRI NIH HHS/United States
- U54HG004563/HG/NHGRI NIH HHS/United States
- R01 HG005085/HG/NHGRI NIH HHS/United States
- U01 HG004571/HG/NHGRI NIH HHS/United States
- U41 HG004568/HG/NHGRI NIH HHS/United States
- R01HG003700/HG/NHGRI NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- 095908/WT_/Wellcome Trust/United Kingdom
- R01 DK065806/DK/NIDDK NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- RC2HG005679/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- U54HG004570/HG/NHGRI NIH HHS/United States
- R01 HG003700/HG/NHGRI NIH HHS/United States
- U01 HG004695/HG/NHGRI NIH HHS/United States
- U54HG004555/HG/NHGRI NIH HHS/United States
- U01 HG004561/HG/NHGRI NIH HHS/United States
- U01HG004571/HG/NHGRI NIH HHS/United States
- U54 HG004555/HG/NHGRI NIH HHS/United States
- R01 HG004456/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
