

In vitro analysis of the role of replication protein A (RPA) and RPA phosphorylation in ATR-mediated checkpoint signaling
- PMID: 22948311
- PMCID: PMC3476280
- DOI: 10.1074/jbc.M112.407825
In vitro analysis of the role of replication protein A (RPA) and RPA phosphorylation in ATR-mediated checkpoint signaling
Abstract
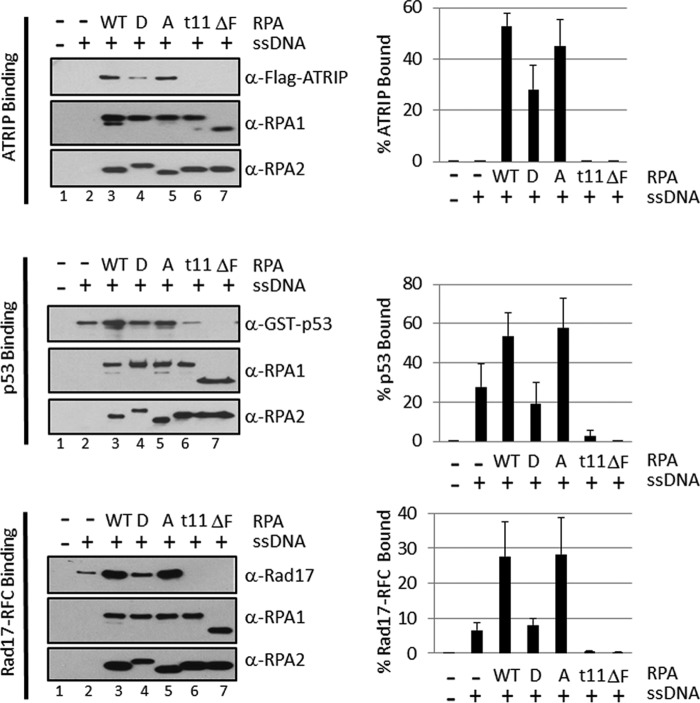
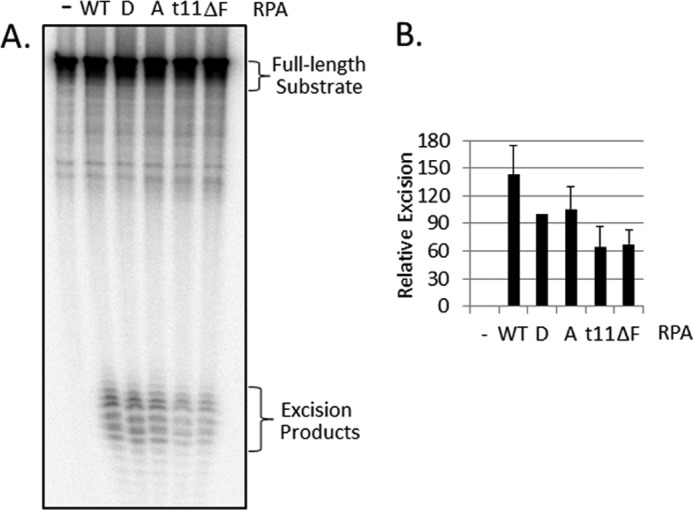
Replication protein A (RPA) plays essential roles in DNA metabolism, including replication, checkpoint, and repair. Recently, we described an in vitro system in which the phosphorylation of human Chk1 kinase by ATR (ataxia telangiectasia mutated and Rad3-related) is dependent on RPA bound to single-stranded DNA. Here, we report that phosphorylation of other ATR targets, p53 and Rad17, has the same requirements and that RPA is also phosphorylated in this system. At high p53 or Rad17 concentrations, RPA phosphorylation is inhibited and, in this system, RPA with phosphomimetic mutations cannot support ATR kinase function, whereas a non-phosphorylatable RPA mutant exhibits full activity. Phosphorylation of these ATR substrates depends on the recruitment of ATR and the substrates by RPA to the RPA-ssDNA complex. Finally, mutant RPAs lacking checkpoint function exhibit essentially normal activity in nucleotide excision repair, revealing RPA separation of function for checkpoint and excision repair.
Figures

Similar articles
-
Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress.Nucleic Acids Res. 2012 Nov;40(21):10780-94. doi: 10.1093/nar/gks849. Epub 2012 Sep 12. Nucleic Acids Res. 2012. PMID: 22977173 Free PMC article.
-
Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling.Proc Natl Acad Sci U S A. 2010 Aug 3;107(31):13660-5. doi: 10.1073/pnas.1007856107. Epub 2010 Jun 28. Proc Natl Acad Sci U S A. 2010. PMID: 20616048 Free PMC article.
-
Phosphorylation of replication protein A by S-phase checkpoint kinases.DNA Repair (Amst). 2006 Mar 7;5(3):369-80. doi: 10.1016/j.dnarep.2005.11.007. Epub 2006 Jan 18. DNA Repair (Amst). 2006. PMID: 16412704
-
Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses.J Cell Physiol. 2006 Aug;208(2):267-73. doi: 10.1002/jcp.20622. J Cell Physiol. 2006. PMID: 16523492 Free PMC article. Review.
-
The ATR barrier to replication-born DNA damage.DNA Repair (Amst). 2010 Dec 10;9(12):1249-55. doi: 10.1016/j.dnarep.2010.09.012. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036674 Free PMC article. Review.
Cited by
-
RHINO forms a stoichiometric complex with the 9-1-1 checkpoint clamp and mediates ATR-Chk1 signaling.Cell Cycle. 2015;14(1):99-108. doi: 10.4161/15384101.2014.967076. Cell Cycle. 2015. PMID: 25602520 Free PMC article.
-
The Nuclear Proteins TP73 and CUL4A Confer Resistance to Cytarabine by Induction of Translesion DNA Synthesis via Mono-ubiquitination of PCNA.Hemasphere. 2022 Apr 26;6(5):e0708. doi: 10.1097/HS9.0000000000000708. eCollection 2022 May. Hemasphere. 2022. PMID: 35519003 Free PMC article.
-
LIM Protein Ajuba associates with the RPA complex through direct cell cycle-dependent interaction with the RPA70 subunit.Sci Rep. 2018 Jun 22;8(1):9536. doi: 10.1038/s41598-018-27919-8. Sci Rep. 2018. PMID: 29934626 Free PMC article.
-
Translesion DNA Synthesis in Cancer: Molecular Mechanisms and Therapeutic Opportunities.Chem Res Toxicol. 2017 Nov 20;30(11):1942-1955. doi: 10.1021/acs.chemrestox.7b00157. Epub 2017 Sep 28. Chem Res Toxicol. 2017. PMID: 28841374 Free PMC article. Review.
-
A tale of two cities: A tribute to Aziz Sancar's Nobel Prize in Chemistry for his molecular characterization of NER.DNA Repair (Amst). 2016 Jan;37:A3-A13. doi: 10.1016/j.dnarep.2015.12.002. DNA Repair (Amst). 2016. PMID: 26861185 Free PMC article. No abstract available.
References
-
- Wold M. S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 - PubMed
-
- Wang Y., Putnam C. D., Kane M. F., Zhang W., Edelmann L., Russell R., Carrión D. V., Chin L., Kucherlapati R., Kolodner R. D., Edelmann W. (2005) Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability, and cancer in mice. Nat. Genet. 37, 750–755 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
