

The inflammation highway: metabolism accelerates inflammatory traffic in obesity
- PMID: 22889225
- PMCID: PMC3422768
- DOI: 10.1111/j.1600-065X.2012.01151.x
The inflammation highway: metabolism accelerates inflammatory traffic in obesity
Abstract
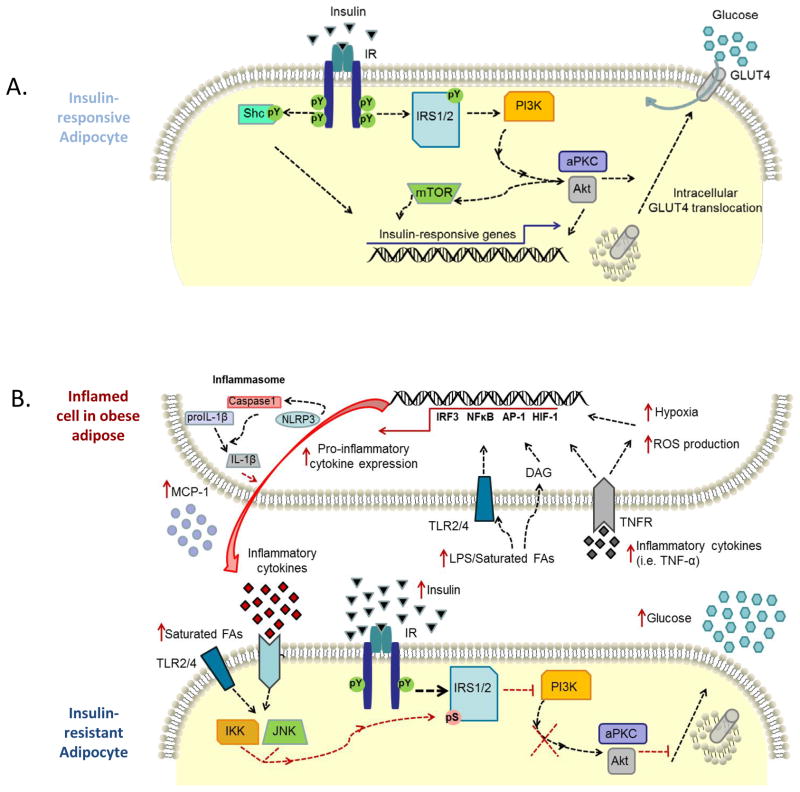
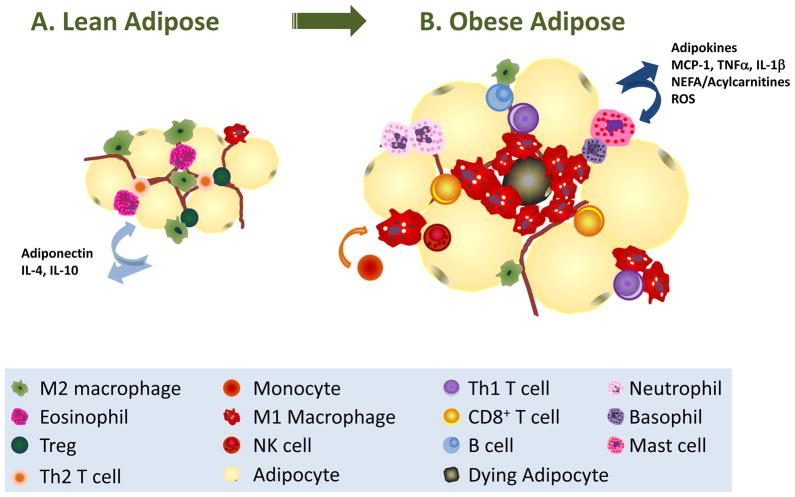
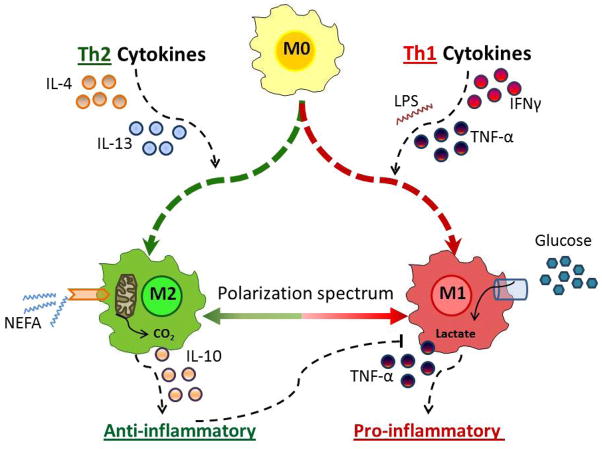
As humans evolved, perhaps the two strongest selection determinants of survival were a robust immune response able to clear bacterial, viral, and parasitic infection and an ability to efficiently store nutrients to survive times when food sources were scarce. These traits are not mutually exclusive. It is now apparent that critical proteins necessary for regulating energy metabolism, such as peroxisome proliferator-activated receptors, Toll-like receptors, and fatty acid-binding proteins, also act as links between nutrient metabolism and inflammatory pathway activation in immune cells. Obesity in humans is a symptom of energy imbalance: the scale has been tipped such that energy intake exceeds energy output and may be a result, in part, of evolutionary selection toward a phenotype characterized by efficient energy storage. As discussed in this review, obesity is a state of low-grade, chronic inflammation that promotes the development of insulin resistance and diabetes. Ironically, the formation of systemic and/or local, tissue-specific insulin resistance upon inflammatory cell activation may actually be a protective mechanism that co-evolved to repartition energy sources within the body during times of stress during infection. However, the point has been reached where a once beneficial adaptive trait has become detrimental to the health of the individual and an immense public health and economic burden. This article reviews the complex relationship between obesity, insulin resistance/diabetes, and inflammation, and although the liver, brain, pancreas, muscle, and other tissues are relevant, we focus specifically on how the obese adipose microenvironment can promote immune cell influx and sustain damaging inflammation that can lead to the onset of insulin resistance and diabetes. Finally, we address how substrate metabolism may regulate the immune response and discuss how fuel uptake and metabolism may be a targetable approach to limit or abrogate obesity-induced inflammation.
© 2012 John Wiley & Sons A/S.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
Similar articles
-
Adipose tissue macrophages: going off track during obesity.Diabetologia. 2016 May;59(5):879-94. doi: 10.1007/s00125-016-3904-9. Epub 2016 Mar 3. Diabetologia. 2016. PMID: 26940592 Free PMC article. Review.
-
Regulation of metabolism by the innate immune system.Nat Rev Endocrinol. 2016 Jan;12(1):15-28. doi: 10.1038/nrendo.2015.189. Epub 2015 Nov 10. Nat Rev Endocrinol. 2016. PMID: 26553134 Review.
-
Effect of energy restriction and physical exercise intervention on phenotypic flexibility as examined by transcriptomics analyses of mRNA from adipose tissue and whole body magnetic resonance imaging.Physiol Rep. 2016 Nov;4(21):e13019. doi: 10.14814/phy2.13019. Physiol Rep. 2016. PMID: 27821717 Free PMC article. Clinical Trial.
-
CCR7 Maintains Nonresolving Lymph Node and Adipose Inflammation in Obesity.Diabetes. 2016 Aug;65(8):2268-81. doi: 10.2337/db15-1689. Epub 2016 May 3. Diabetes. 2016. PMID: 27207557 Free PMC article.
-
Recent advances in the relationship between obesity, inflammation, and insulin resistance.Eur Cytokine Netw. 2006 Mar;17(1):4-12. Eur Cytokine Netw. 2006. PMID: 16613757 Review.
Cited by
-
Regulation of visfatin by microbial and biomechanical signals in PDL cells.Clin Oral Investig. 2014 Jan;18(1):171-8. doi: 10.1007/s00784-013-0935-1. Epub 2013 Feb 13. Clin Oral Investig. 2014. PMID: 23404558
-
Distinct Adipose Depots from Mice Differentially Respond to a High-Fat, High-Salt Diet.J Nutr. 2016 Jun;146(6):1189-96. doi: 10.3945/jn.115.227496. Epub 2016 May 4. J Nutr. 2016. PMID: 27146921 Free PMC article.
-
The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background.Gastroenterol Res Pract. 2016;2016:2862173. doi: 10.1155/2016/2862173. Epub 2016 May 9. Gastroenterol Res Pract. 2016. PMID: 27247565 Free PMC article. Review.
-
Innate immune signaling and gut-liver interactions in non-alcoholic fatty liver disease.Hepatobiliary Surg Nutr. 2014 Dec;3(6):377-85. doi: 10.3978/j.issn.2304-3881.2014.12.04. Hepatobiliary Surg Nutr. 2014. PMID: 25568861 Free PMC article. Review.
-
NLRP3 Inflammasome in Inflammation and Metabolism: Identifying Novel Roles in Postburn Adipose Dysfunction.Endocrinology. 2020 Sep 1;161(9):bqaa116. doi: 10.1210/endocr/bqaa116. Endocrinology. 2020. PMID: 32790834 Free PMC article. Review.
References
-
- Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. - PubMed
-
- Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. Jama. 2006;295:1549–1555. - PubMed
-
- Organization WH. Obesity and Overweight. 2012
-
- Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62. - PubMed
-
- Grundy SM. What is the contribution of obesity to the metabolic syndrome? Endocrinol Metab Clin North Am. 2004;33:267–282. table of contents. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
