

Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry
- PMID: 22855500
- PMCID: PMC3457153
- DOI: 10.1128/JVI.00750-12
Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry
Abstract
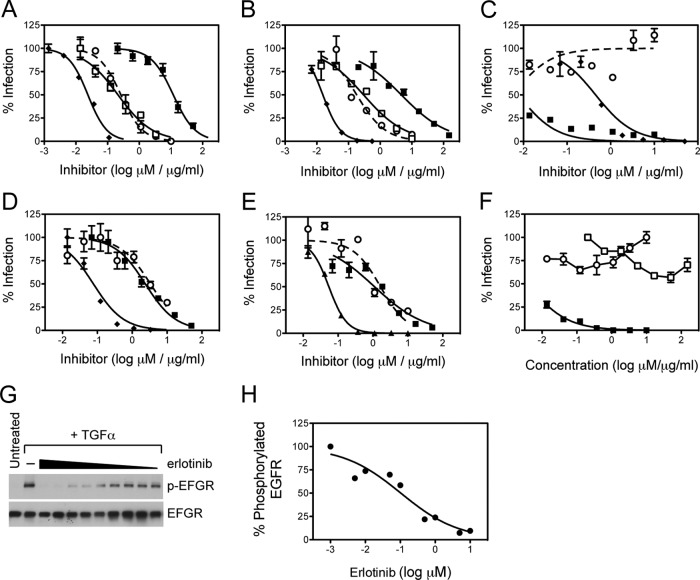
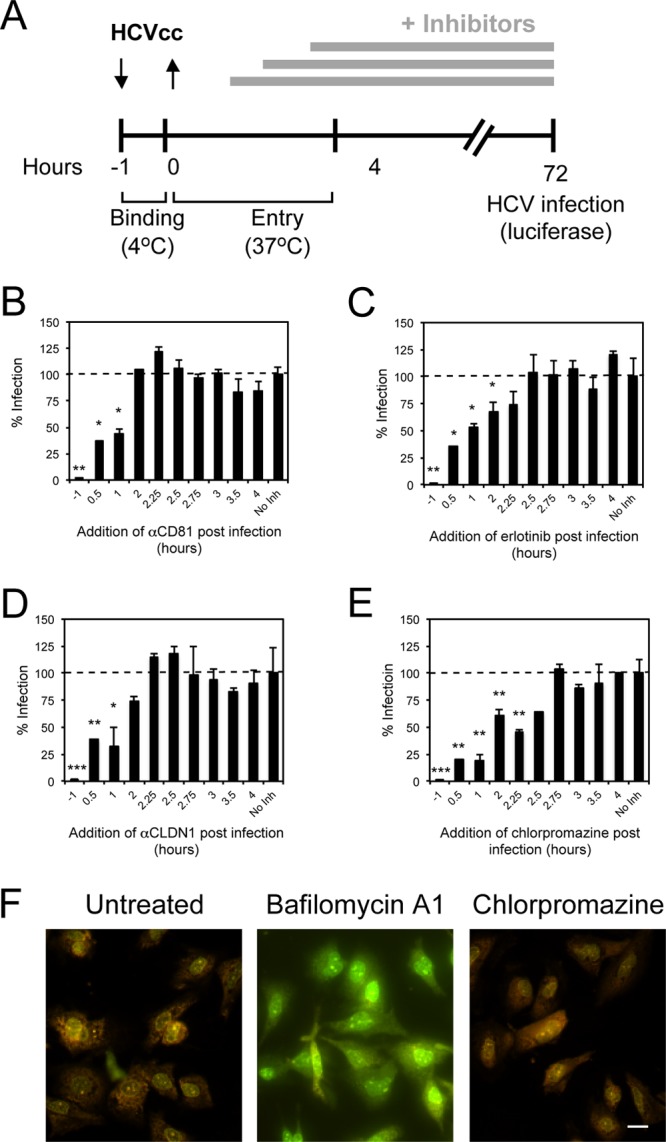
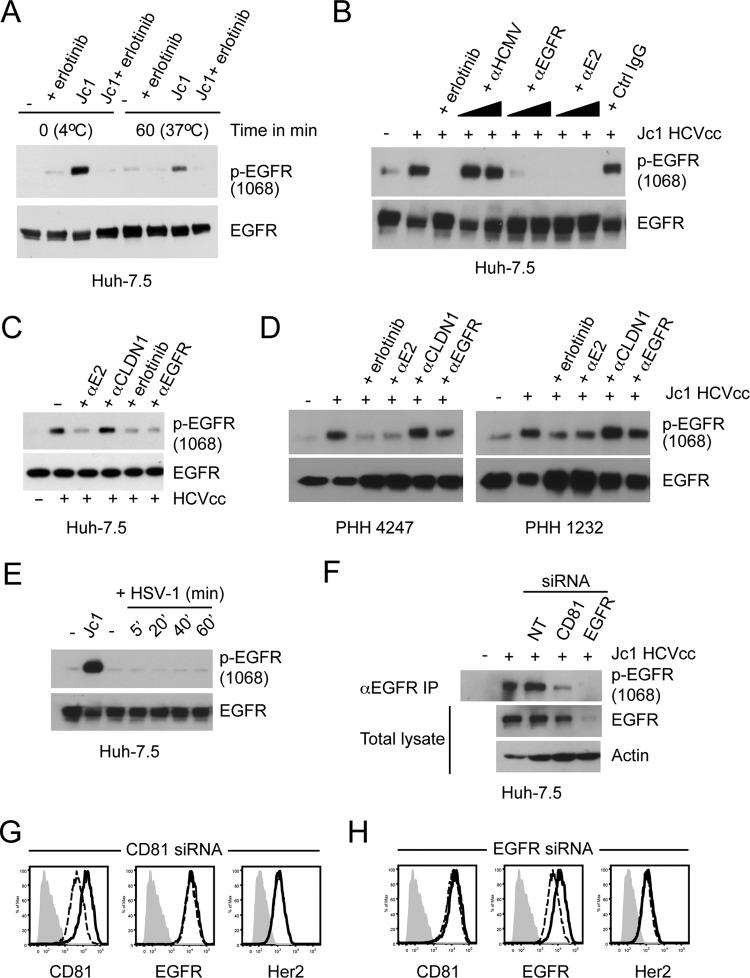
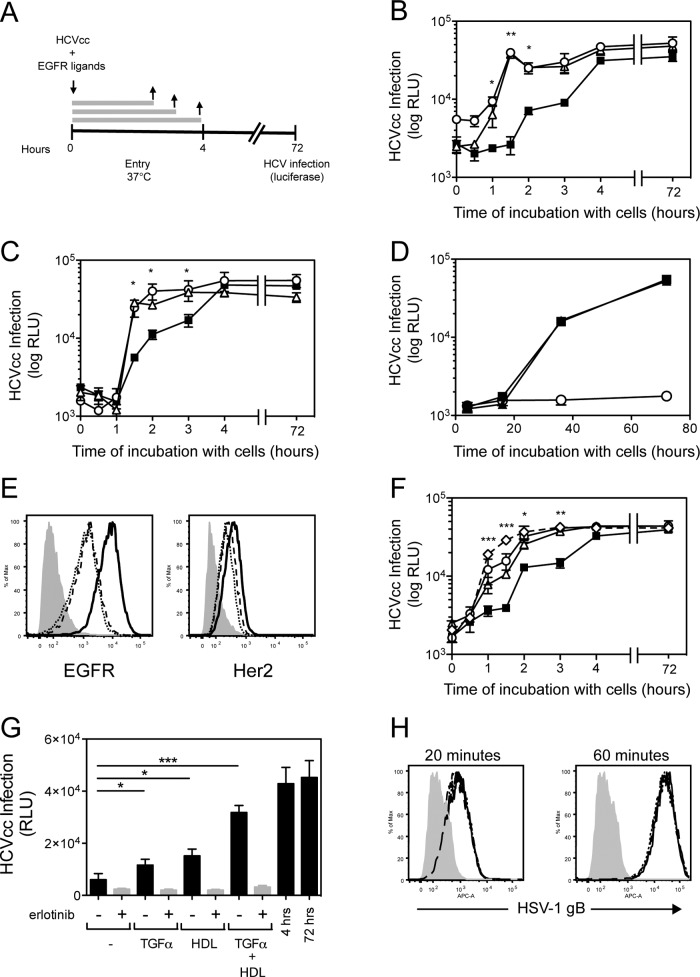
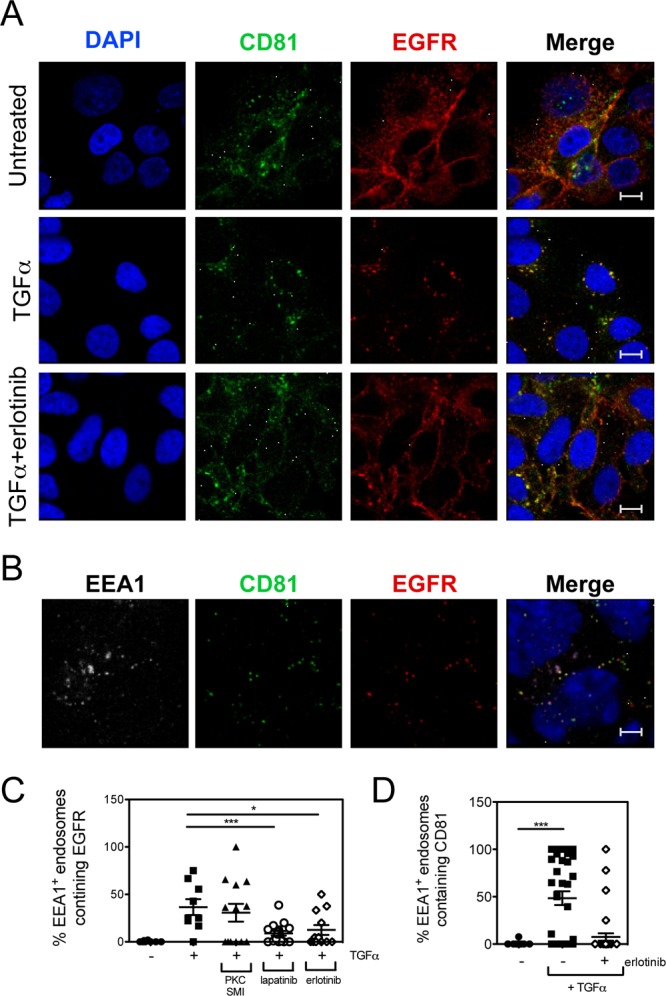
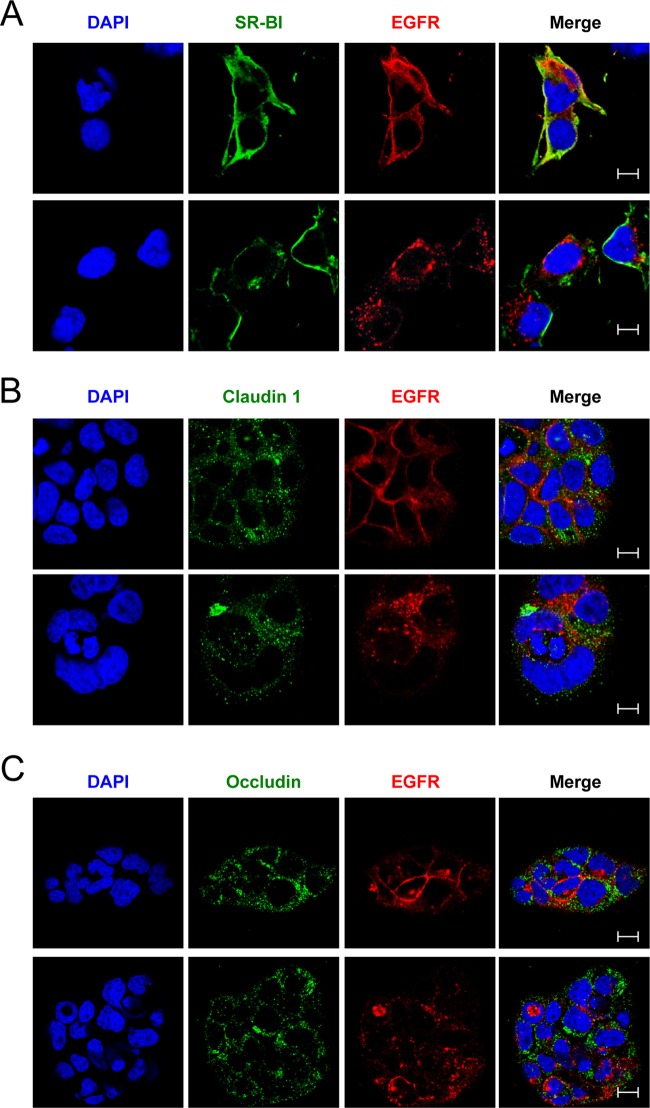
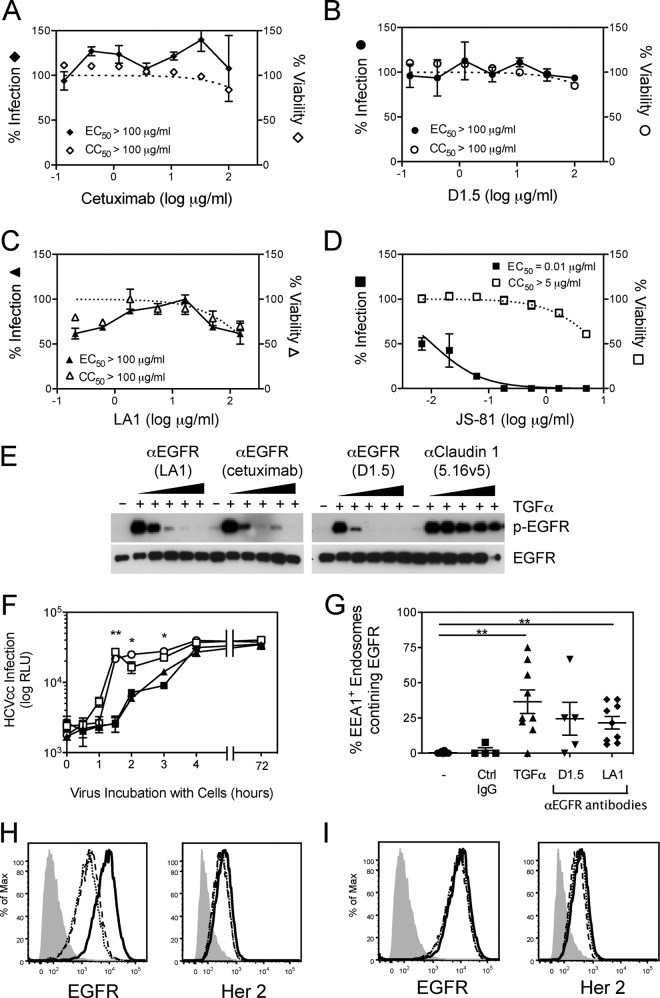
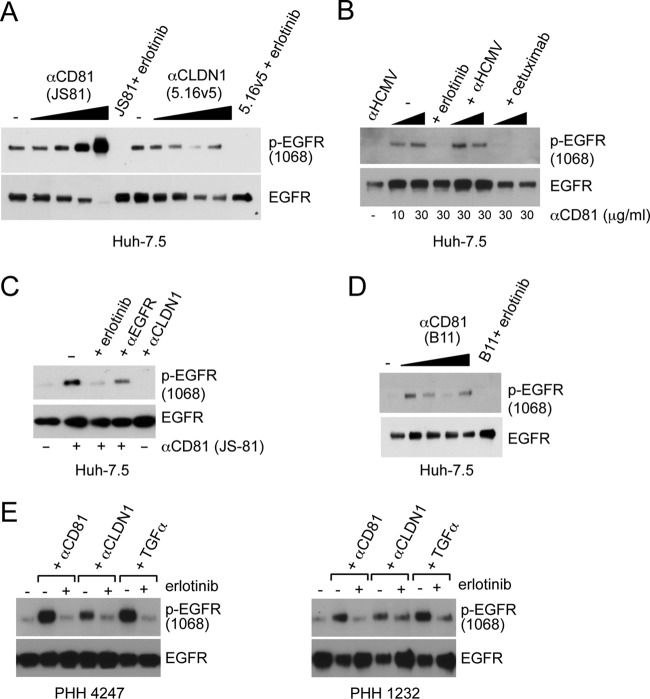
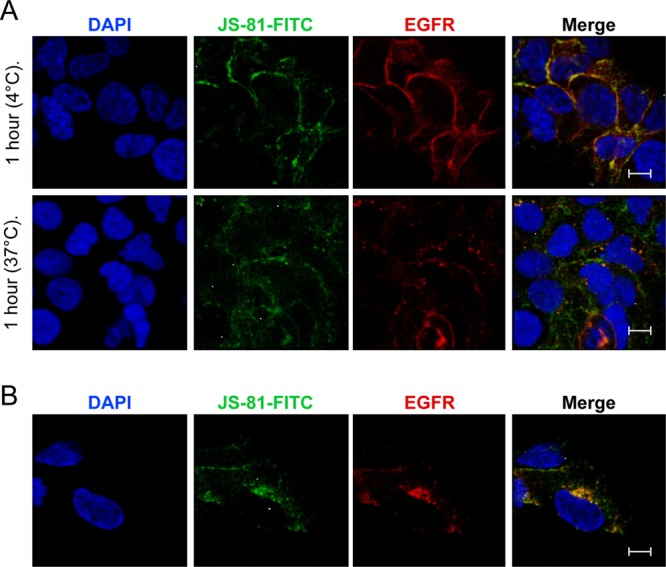
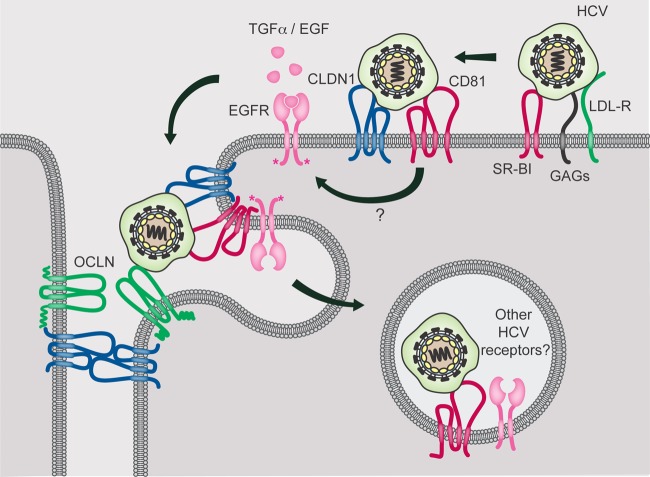
While epidermal growth factor receptor (EGFR) has been shown to be important in the entry process for multiple viruses, including hepatitis C virus (HCV), the molecular mechanisms by which EGFR facilitates HCV entry are not well understood. Using the infectious cell culture HCV model (HCVcc), we demonstrate that the binding of HCVcc particles to human hepatocyte cells induces EGFR activation that is dependent on interactions between HCV and CD81 but not claudin 1. EGFR activation can also be induced by antibody mediated cross-linking of CD81. In addition, EGFR ligands that enhance the kinetics of HCV entry induce EGFR internalization and colocalization with CD81. While EGFR kinase inhibitors inhibit HCV infection primarily by preventing EGFR endocytosis, antibodies that block EGFR ligand binding or inhibitors of EGFR downstream signaling have no effect on HCV entry. These data demonstrate that EGFR internalization is critical for HCV entry and identify a hitherto-unknown association between CD81 and EGFR.
Figures
Similar articles
-
Hepatitis C virus induces CD81 and claudin-1 endocytosis.J Virol. 2012 Apr;86(8):4305-16. doi: 10.1128/JVI.06996-11. Epub 2012 Feb 8. J Virol. 2012. PMID: 22318146 Free PMC article.
-
HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex.Cell Host Microbe. 2013 Mar 13;13(3):302-13. doi: 10.1016/j.chom.2013.02.006. Cell Host Microbe. 2013. PMID: 23498955
-
Single Particle Imaging of Polarized Hepatoma Organoids upon Hepatitis C Virus Infection Reveals an Ordered and Sequential Entry Process.Cell Host Microbe. 2018 Mar 14;23(3):382-394.e5. doi: 10.1016/j.chom.2018.02.005. Cell Host Microbe. 2018. PMID: 29544098 Free PMC article.
-
Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion.Cold Spring Harb Perspect Med. 2020 Feb 3;10(2):a036830. doi: 10.1101/cshperspect.a036830. Cold Spring Harb Perspect Med. 2020. PMID: 31427285 Free PMC article. Review.
-
Hepatitis C virus entry and the tetraspanin CD81.Biochem Soc Trans. 2011 Apr;39(2):532-6. doi: 10.1042/BST0390532. Biochem Soc Trans. 2011. PMID: 21428934 Review.
Cited by
-
Kinase Inhibitors as Underexplored Antiviral Agents.J Med Chem. 2022 Jan 27;65(2):935-954. doi: 10.1021/acs.jmedchem.1c00302. Epub 2021 May 10. J Med Chem. 2022. PMID: 33970631 Free PMC article. Review.
-
Role of Virus-Induced EGFR Trafficking in Proviral Functions.Biomolecules. 2023 Dec 9;13(12):1766. doi: 10.3390/biom13121766. Biomolecules. 2023. PMID: 38136637 Free PMC article. Review.
-
Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process.Int J Mol Sci. 2020 Mar 18;21(6):2091. doi: 10.3390/ijms21062091. Int J Mol Sci. 2020. PMID: 32197477 Free PMC article. Review.
-
Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge.MAbs. 2014;6(5):1255-64. doi: 10.4161/mabs.29809. Epub 2014 Oct 30. MAbs. 2014. PMID: 25517310 Free PMC article.
-
Repurposing clinically available drugs and therapies for pathogenic targets to combat SARS-CoV-2.MedComm (2020). 2023 May 14;4(3):e254. doi: 10.1002/mco2.254. eCollection 2023 Jun. MedComm (2020). 2023. PMID: 37193304 Free PMC article. Review.
References
-
- Bradley DW. 2000. Studies of non-A, non-B hepatitis and characterization of the hepatitis C virus in chimpanzees. Curr. Top. Microbiol. Immunol. 242:1–23 - PubMed
-
- Burlone ME, Budkowska A. 2009. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J. Gen. Virol. 90:1055–1070 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
