

Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood
- PMID: 22851595
- PMCID: PMC3428952
- DOI: 10.1084/jem.20111316
Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood
Abstract
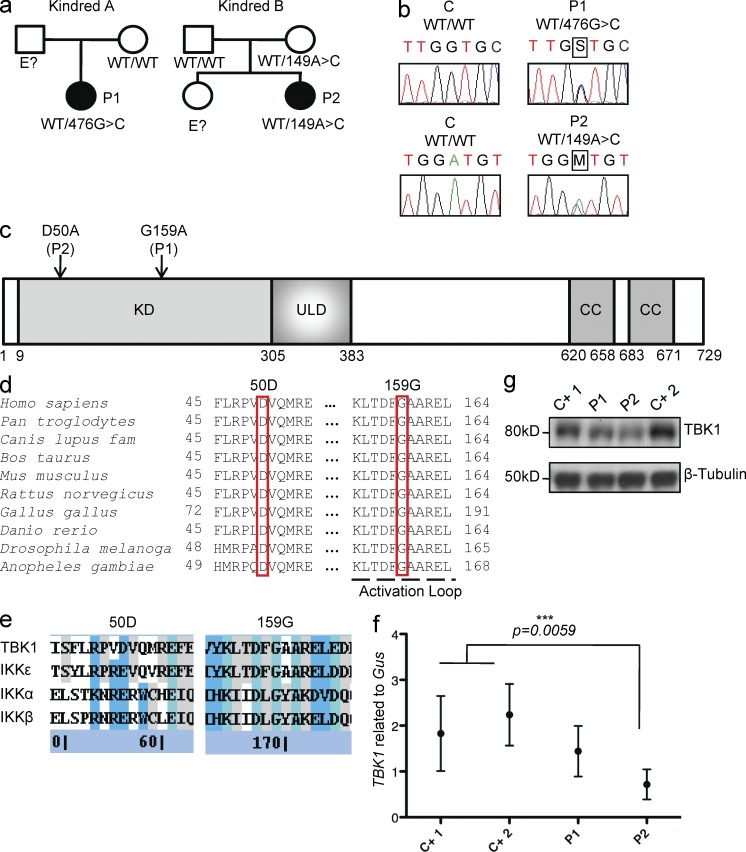
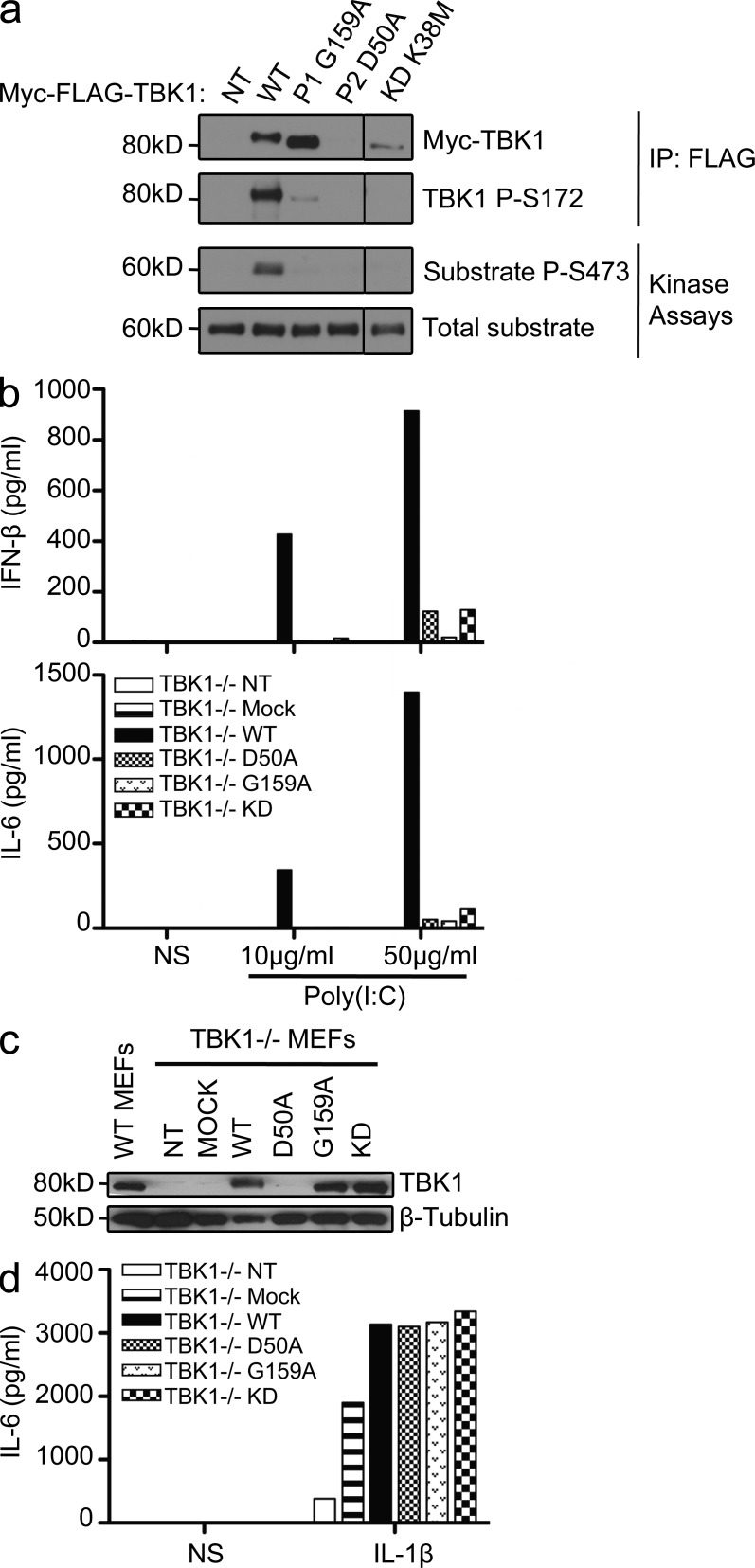
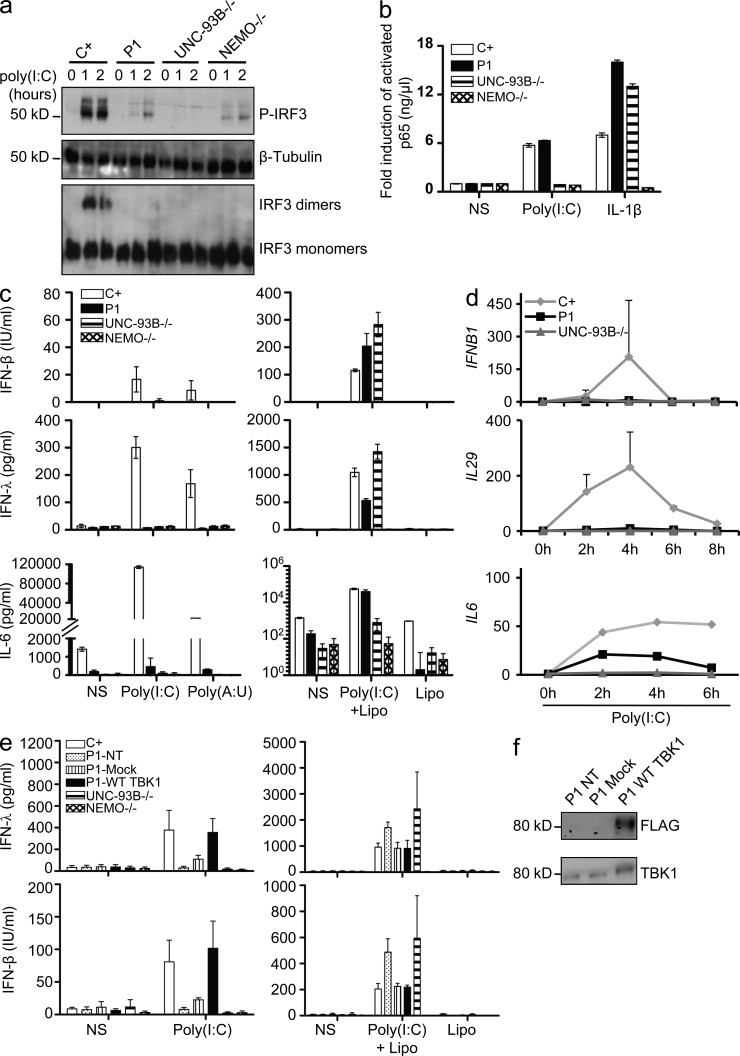
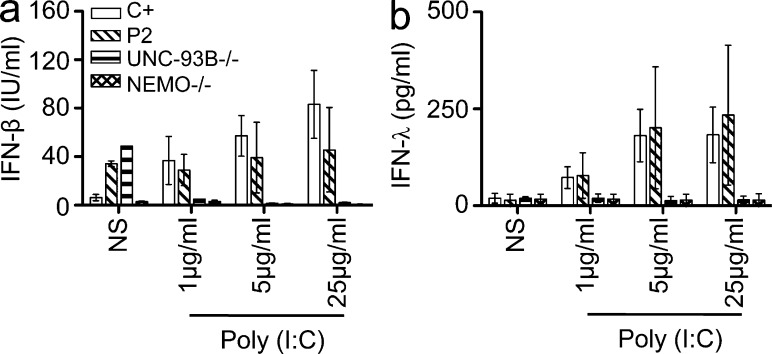
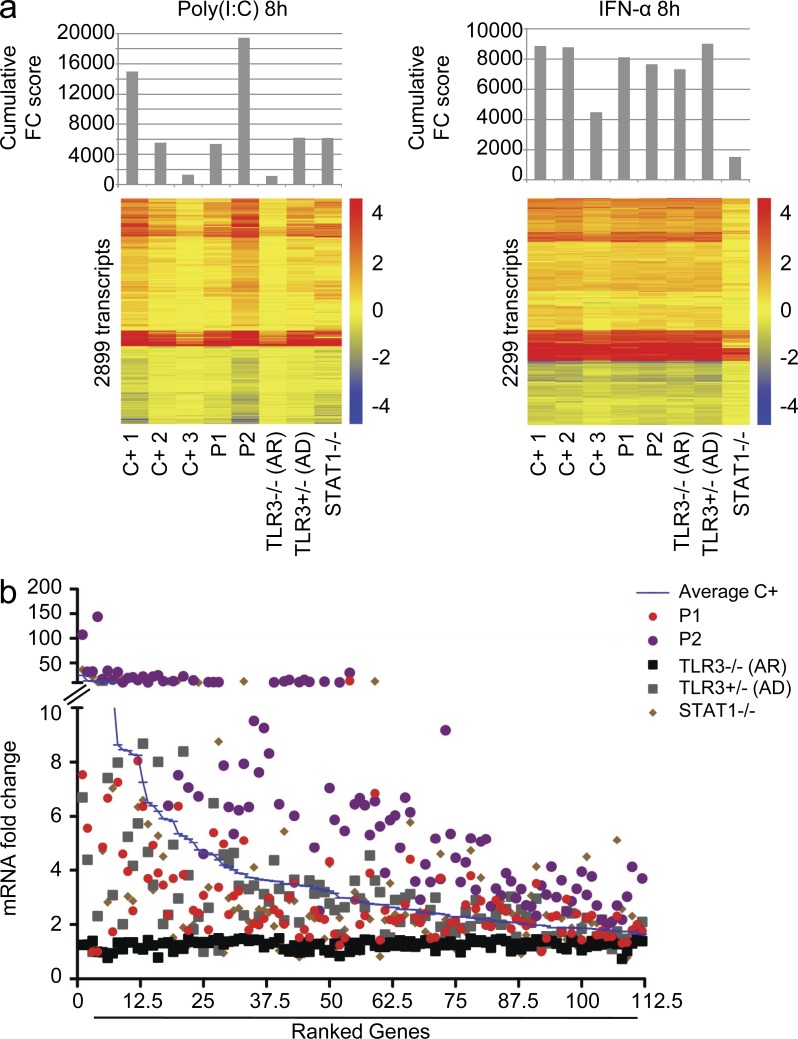
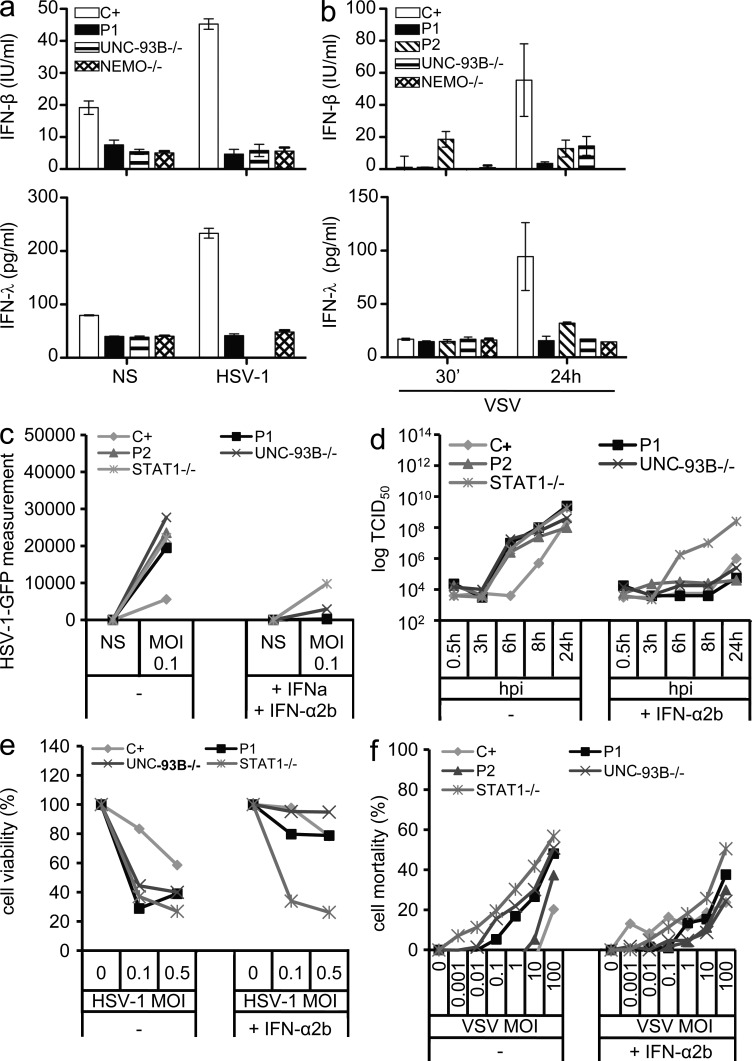
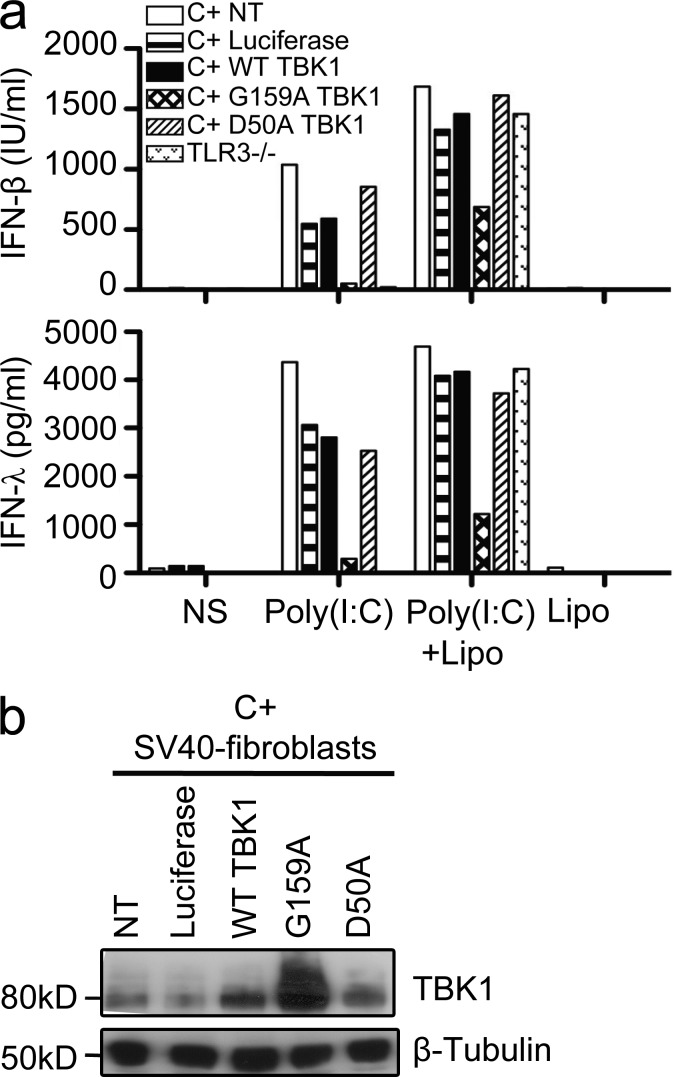
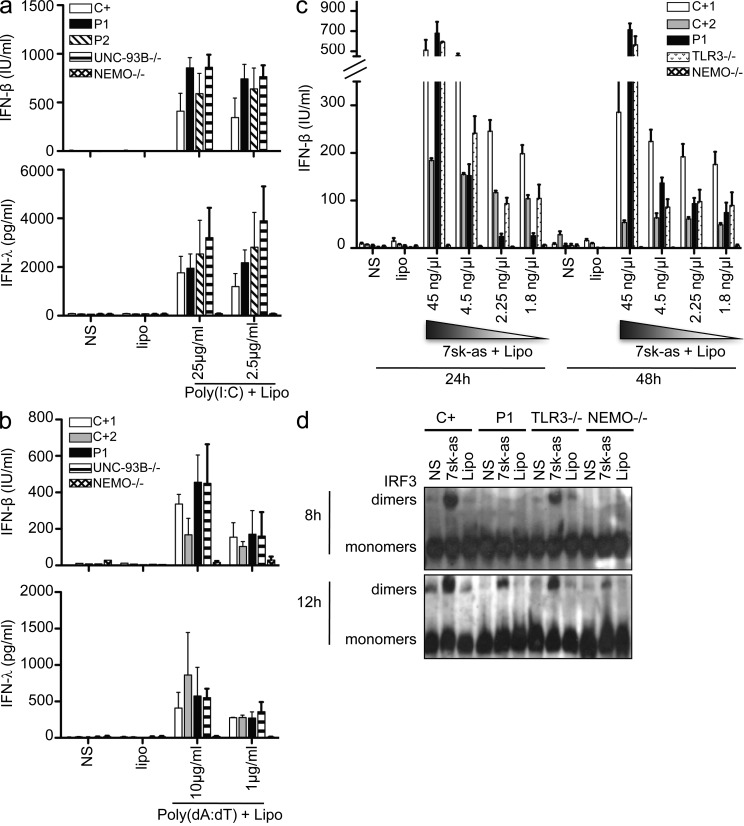
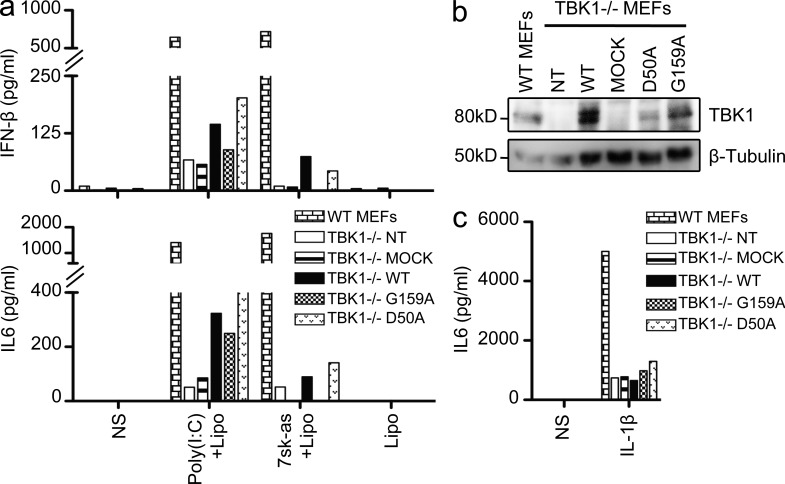
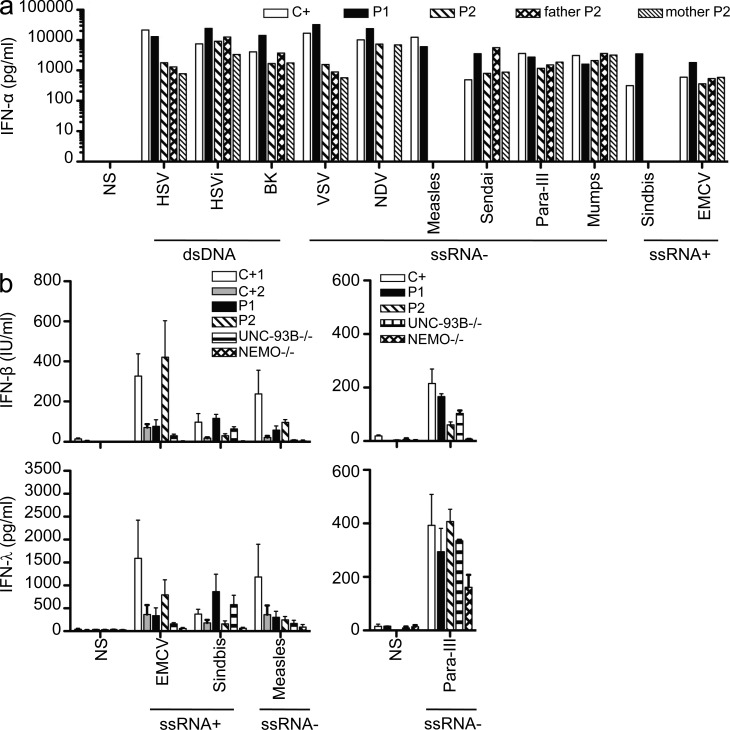
Childhood herpes simplex virus-1 (HSV-1) encephalitis (HSE) may result from single-gene inborn errors of TLR3 immunity. TLR3-dependent induction of IFN-α/β or IFN-λ is crucial for protective immunity against primary HSV-1 infection in the central nervous system (CNS). We describe here two unrelated children with HSE carrying different heterozygous mutations (D50A and G159A) in TBK1, the gene encoding TANK-binding kinase 1, a kinase at the crossroads of multiple IFN-inducing signaling pathways. Both mutant TBK1 alleles are loss-of-function but through different mechanisms: protein instability (D50A) or a loss of kinase activity (G159A). Both are also associated with an autosomal-dominant (AD) trait but by different mechanisms: haplotype insufficiency (D50A) or negative dominance (G159A). A defect in polyinosinic-polycytidylic acid-induced TLR3 responses can be detected in fibroblasts heterozygous for G159A but not for D50A TBK1. Nevertheless, viral replication and cell death rates caused by two TLR3-dependent viruses (HSV-1 and vesicular stomatitis virus) were high in fibroblasts from both patients, and particularly so in G159A TBK1 fibroblasts. These phenotypes were rescued equally well by IFN-α2b. Moreover, the IFN responses to the TLR3-independent agonists and viruses tested were maintained in both patients' peripheral blood mononuclear cells and fibroblasts. The narrow, partial cellular phenotype thus accounts for the clinical phenotype of these patients being limited to HSE. These data identify AD partial TBK1 deficiency as a new genetic etiology of childhood HSE, indicating that TBK1 is essential for the TLR3- and IFN-dependent control of HSV-1 in the CNS.
Figures
Similar articles
-
Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity.J Exp Med. 2011 Sep 26;208(10):2083-98. doi: 10.1084/jem.20101568. Epub 2011 Sep 12. J Exp Med. 2011. PMID: 21911422 Free PMC article.
-
Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells.Nature. 2012 Nov 29;491(7426):769-73. doi: 10.1038/nature11583. Epub 2012 Oct 28. Nature. 2012. PMID: 23103873 Free PMC article.
-
NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus.J Allergy Clin Immunol. 2011 Sep;128(3):610-7.e1-4. doi: 10.1016/j.jaci.2011.04.059. Epub 2011 Jul 1. J Allergy Clin Immunol. 2011. PMID: 21722947 Free PMC article.
-
Mendelian predisposition to herpes simplex encephalitis.Handb Clin Neurol. 2013;112:1091-7. doi: 10.1016/B978-0-444-52910-7.00027-1. Handb Clin Neurol. 2013. PMID: 23622315 Review.
-
Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity.Hum Genet. 2020 Jun;139(6-7):911-918. doi: 10.1007/s00439-020-02127-5. Epub 2020 Feb 10. Hum Genet. 2020. PMID: 32040615 Free PMC article. Review.
Cited by
-
The immunobiology of herpes simplex virus encephalitis and post-viral autoimmunity.Brain. 2024 Apr 4;147(4):1130-1148. doi: 10.1093/brain/awad419. Brain. 2024. PMID: 38092513 Free PMC article. Review.
-
Targeting TANK-binding kinase 1 (TBK1) in cancer.Expert Opin Ther Targets. 2020 Nov;24(11):1065-1078. doi: 10.1080/14728222.2020.1826929. Epub 2020 Oct 5. Expert Opin Ther Targets. 2020. PMID: 32962465 Free PMC article. Review.
-
General Pathophysiology of Astroglia.Adv Exp Med Biol. 2019;1175:149-179. doi: 10.1007/978-981-13-9913-8_7. Adv Exp Med Biol. 2019. PMID: 31583588 Free PMC article. Review.
-
Effects of ALS-associated TANK binding kinase 1 mutations on protein-protein interactions and kinase activity.Proc Natl Acad Sci U S A. 2019 Dec 3;116(49):24517-24526. doi: 10.1073/pnas.1915732116. Epub 2019 Nov 20. Proc Natl Acad Sci U S A. 2019. PMID: 31748271 Free PMC article.
-
Severe COVID-19 represents an undiagnosed primary immunodeficiency in a high proportion of infected individuals.Clin Transl Immunology. 2022 Apr 17;11(4):e1365. doi: 10.1002/cti2.1365. eCollection 2022. Clin Transl Immunology. 2022. PMID: 35444807 Free PMC article. Review.
References
-
- Abel L., Plancoulaine S., Jouanguy E., Zhang S.Y., Mahfoufi N., Nicolas N., Sancho-Shimizu V., Alcaïs A., Guo Y., Cardon A., et al. 2010. Age-dependent Mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J. Pediatr. 157:623–629: 629: e1 10.1016/j.jpeds.2010.04.020 - DOI - PubMed
-
- Audry M., Ciancanelli M., Yang K., Cobat A., Chang H.H., Sancho-Shimizu V., Lorenzo L., Niehues T., Reichenbach J., Li X.X., et al. 2011. NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 128:610–617: e1–e4 10.1016/j.jaci.2011.04.059 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
