

The origin and evolution of mutations in acute myeloid leukemia
- PMID: 22817890
- PMCID: PMC3407563
- DOI: 10.1016/j.cell.2012.06.023
The origin and evolution of mutations in acute myeloid leukemia
Abstract
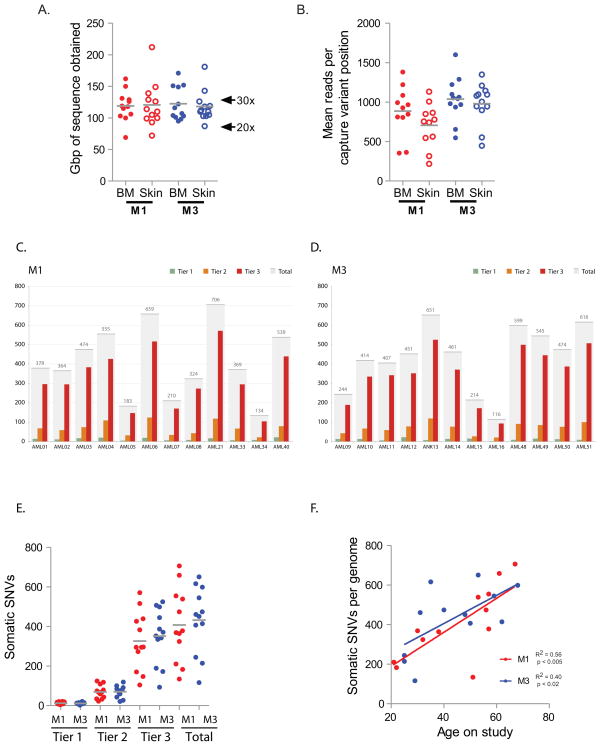
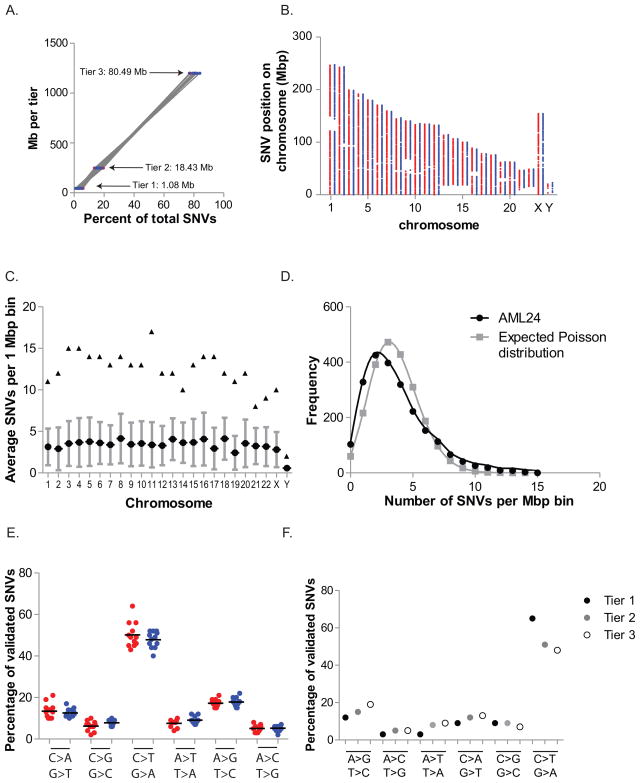
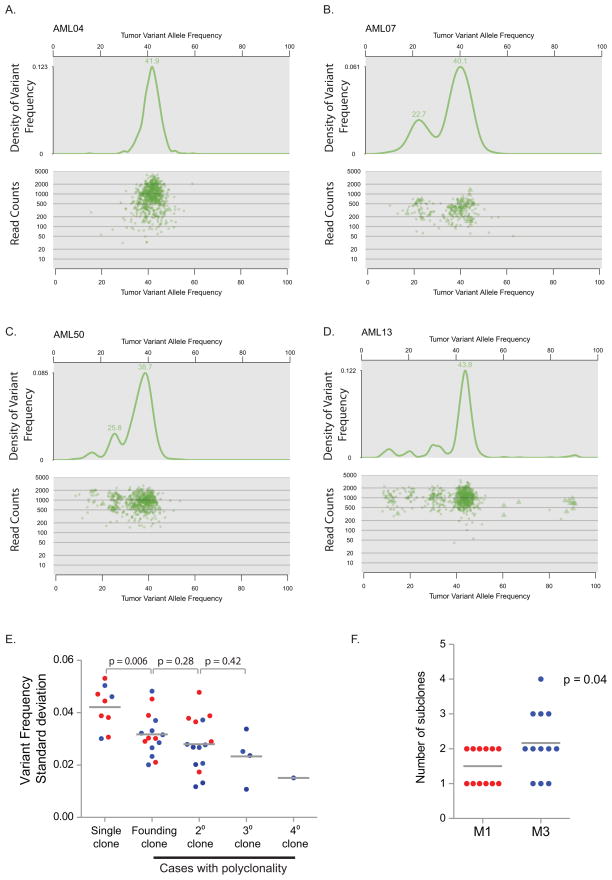
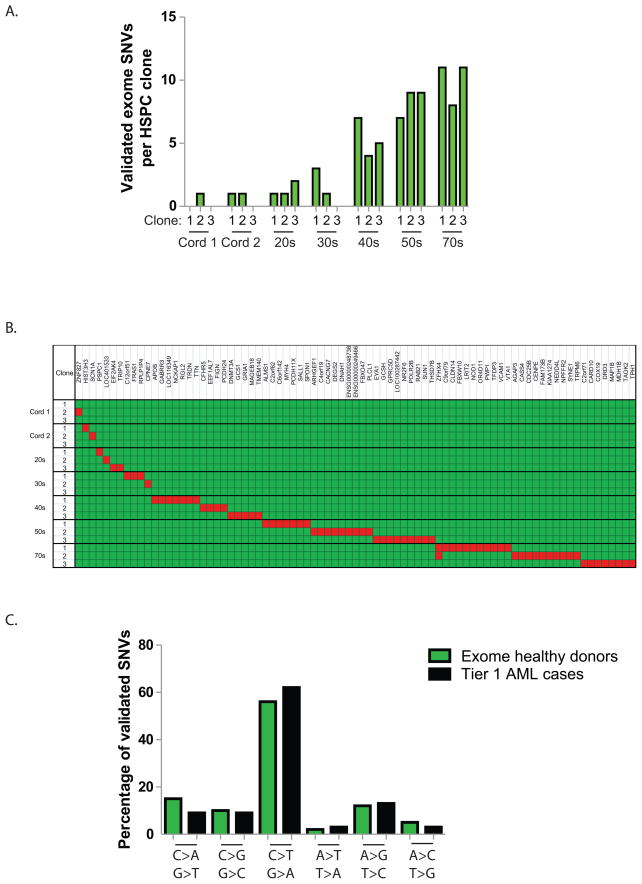
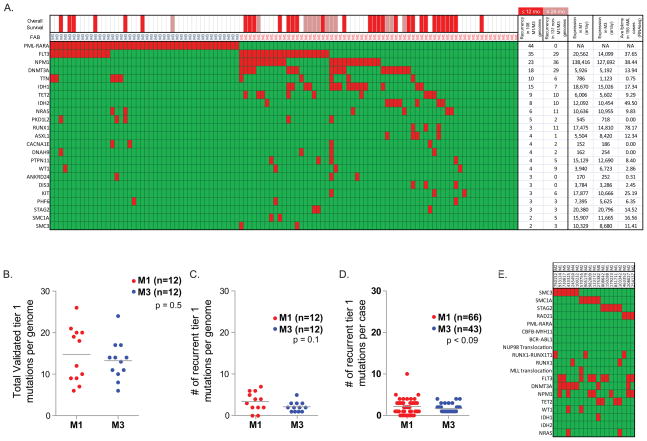
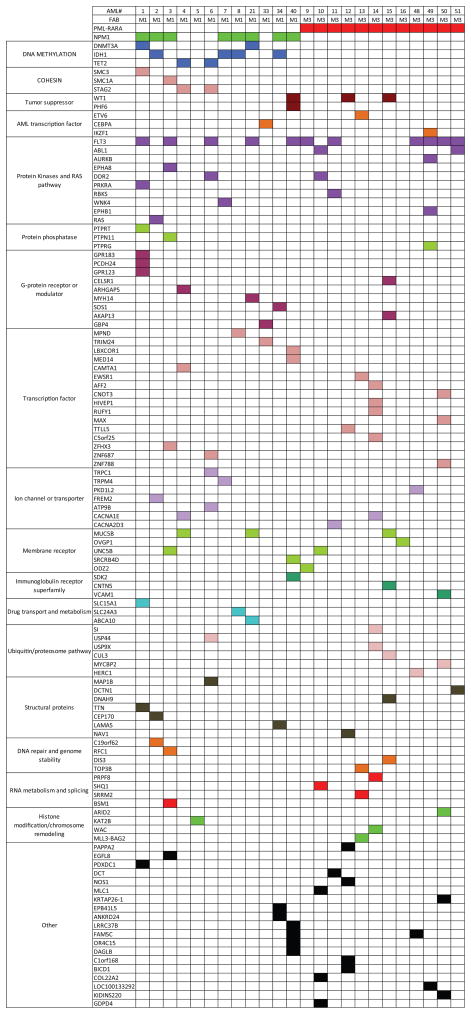
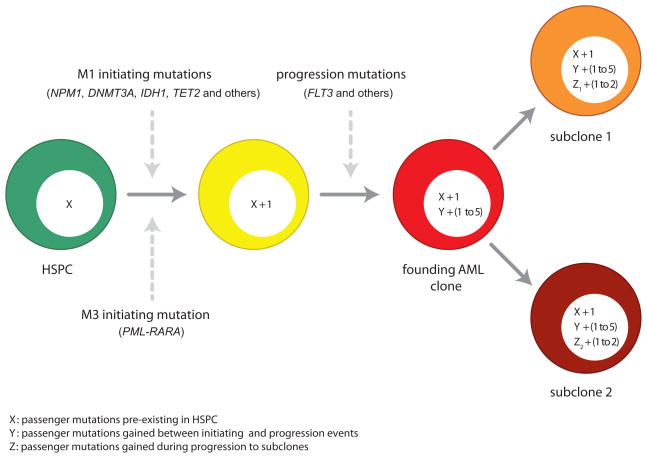
Most mutations in cancer genomes are thought to be acquired after the initiating event, which may cause genomic instability and drive clonal evolution. However, for acute myeloid leukemia (AML), normal karyotypes are common, and genomic instability is unusual. To better understand clonal evolution in AML, we sequenced the genomes of M3-AML samples with a known initiating event (PML-RARA) versus the genomes of normal karyotype M1-AML samples and the exomes of hematopoietic stem/progenitor cells (HSPCs) from healthy people. Collectively, the data suggest that most of the mutations found in AML genomes are actually random events that occurred in HSPCs before they acquired the initiating mutation; the mutational history of that cell is "captured" as the clone expands. In many cases, only one or two additional, cooperating mutations are needed to generate the malignant founding clone. Cells from the founding clone can acquire additional cooperating mutations, yielding subclones that can contribute to disease progression and/or relapse.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Clonal evolution of acute leukemia genomes.Oncogene. 2013 Jan 10;32(2):135-40. doi: 10.1038/onc.2012.48. Epub 2012 Feb 20. Oncogene. 2013. PMID: 22349821 Free PMC article. Review.
-
Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing.Nature. 2012 Jan 11;481(7382):506-10. doi: 10.1038/nature10738. Nature. 2012. PMID: 22237025 Free PMC article.
-
Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia.Sci Transl Med. 2012 Aug 29;4(149):149ra118. doi: 10.1126/scitranslmed.3004315. Sci Transl Med. 2012. PMID: 22932223 Free PMC article.
-
Clonal evolution of acute myeloid leukemia highlighted by latest genome sequencing studies.Oncotarget. 2016 Sep 6;7(36):58586-58594. doi: 10.18632/oncotarget.10850. Oncotarget. 2016. PMID: 27474172 Free PMC article. Review.
-
Monitoring of clonal evolution of double C-KIT exon 17 mutations by Droplet Digital PCR in patients with core-binding factor acute myeloid leukemia.Leuk Res. 2018 Jun;69:89-93. doi: 10.1016/j.leukres.2018.04.013. Epub 2018 Apr 22. Leuk Res. 2018. PMID: 29705537
Cited by
-
How aging influences the gut-bone marrow axis and alters hematopoietic stem cell regulation.Heliyon. 2024 Jun 11;10(12):e32831. doi: 10.1016/j.heliyon.2024.e32831. eCollection 2024 Jun 30. Heliyon. 2024. PMID: 38984298 Free PMC article. Review.
-
Next generation sequencing of acute myeloid leukemia: influencing prognosis.BMC Genomics. 2015;16 Suppl 1(Suppl 1):S5. doi: 10.1186/1471-2164-16-S1-S5. Epub 2015 Jan 15. BMC Genomics. 2015. PMID: 25924101 Free PMC article. Review.
-
Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia.N Engl J Med. 2013 May 30;368(22):2059-74. doi: 10.1056/NEJMoa1301689. Epub 2013 May 1. N Engl J Med. 2013. PMID: 23634996 Free PMC article.
-
OTS167 blocks FLT3 translation and synergizes with FLT3 inhibitors in FLT3 mutant acute myeloid leukemia.Blood Cancer J. 2021 Mar 3;11(3):48. doi: 10.1038/s41408-021-00433-3. Blood Cancer J. 2021. PMID: 33658483 Free PMC article.
-
Clonal Hematopoiesis of Indeterminate Potential in Patients with Solid Tumor Malignancies.Cancer Res. 2022 Nov 15;82(22):4107-4113. doi: 10.1158/0008-5472.CAN-22-0985. Cancer Res. 2022. PMID: 36040522 Free PMC article.
References
-
- Abkowitz JL, Catlin SN, Guttorp P. Evidence that hematopoiesis may be a stochastic process in vivo. Nat Med. 1996;2:190–197. - PubMed
-
- Abkowitz JL, Golinelli D, Harrison DE, Guttorp P. In vivo kinetics of murine hemopoietic stem cells. Blood. 2000;96:3399–3405. - PubMed
-
- Allford S, Grimwade D, Langabeer S, Duprez E, Saurin A, Chatters S, Walker H, Roberts P, Rogers J, Bain B, et al. Identification of the t(15;17) in AML FAB types other than M3: evaluation of the role of molecular screening for the PML/RARalpha rearrangement in newly diagnosed AML. The Medical Research Council (MRC) Adult Leukaemia Working Party. Br J Haematol. 1999;105:198–207. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
