

Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease
- PMID: 22751174
- PMCID: PMC4416477
- DOI: 10.3233/JAD-2012-129037
Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease
Abstract
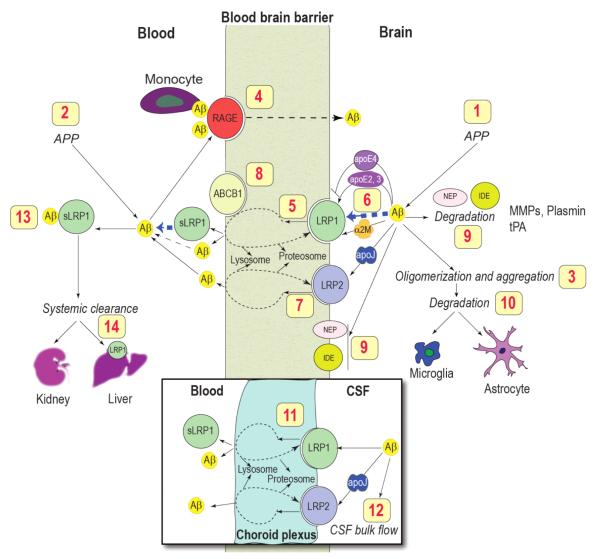
The evidence that neurovascular dysfunction is an integral part of Alzheimer's disease (AD) pathogenesis has continued to emerge in the last decade. Changes in the brain vasculature have been shown to contribute to the onset and progression of the pathological processes associated with AD, such as microvascular reductions, blood brain barrier (BBB) breakdown, and faulty clearance of amyloid β-peptide (Aβ) from the brain. Herein, we review the role of the neurovascular unit and molecular mechanisms in cerebral vascular cells behind the pathogenesis of AD. In particular, we focus on molecular pathways within cerebral vascular cells and the systemic circulation that contribute to BBB dysfunction, brain hypoperfusion, and impaired clearance of Aβ from the brain. We aim to provide a summary of recent research findings implicated in neurovascular defects and faulty Aβ vascular clearance contributing to AD pathogenesis.
Figures

Similar articles
-
Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease.Cold Spring Harb Perspect Med. 2012 Oct 1;2(10):a011452. doi: 10.1101/cshperspect.a011452. Cold Spring Harb Perspect Med. 2012. PMID: 23028132 Free PMC article. Review.
-
Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease.Acta Neuropathol. 2009 Jul;118(1):103-13. doi: 10.1007/s00401-009-0522-3. Epub 2009 Mar 25. Acta Neuropathol. 2009. PMID: 19319544 Free PMC article. Review.
-
The blood brain barrier in Alzheimer's disease.Vascul Pharmacol. 2017 Feb;89:12-18. doi: 10.1016/j.vph.2016.11.008. Epub 2016 Nov 25. Vascul Pharmacol. 2017. PMID: 27894893 Review.
-
Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease.Curr Alzheimer Res. 2007 Apr;4(2):191-7. doi: 10.2174/156720507780362245. Curr Alzheimer Res. 2007. PMID: 17430246 Review.
-
Alzheimer's Disease: The Link Between Amyloid-β and Neurovascular Dysfunction.J Alzheimers Dis. 2020;76(4):1179-1198. doi: 10.3233/JAD-200473. J Alzheimers Dis. 2020. PMID: 32597813 Free PMC article. Review.
Cited by
-
Age-Related Decline in Brain and Hepatic Clearance of Amyloid-Beta is Rectified by the Cholinesterase Inhibitors Donepezil and Rivastigmine in Rats.ACS Chem Neurosci. 2015 May 20;6(5):725-36. doi: 10.1021/acschemneuro.5b00040. Epub 2015 Mar 30. ACS Chem Neurosci. 2015. PMID: 25782004 Free PMC article.
-
Chromosome silencing in vitro reveals trisomy 21 causes cell-autonomous deficits in angiogenesis and early dysregulation in Notch signaling.Cell Rep. 2022 Aug 9;40(6):111174. doi: 10.1016/j.celrep.2022.111174. Cell Rep. 2022. PMID: 35947952 Free PMC article.
-
Plasma Amyloid-β dynamics in late-life major depression: a longitudinal study.Transl Psychiatry. 2022 Jul 28;12(1):301. doi: 10.1038/s41398-022-02077-8. Transl Psychiatry. 2022. PMID: 35902554 Free PMC article.
-
Brain-Derived Exosomal Proteins as Effective Biomarkers for Alzheimer's Disease: A Systematic Review and Meta-Analysis.Biomolecules. 2021 Jul 3;11(7):980. doi: 10.3390/biom11070980. Biomolecules. 2021. PMID: 34356604 Free PMC article.
-
Unveiling the Complex Role of Exosomes in Alzheimer's Disease.J Inflamm Res. 2024 Jun 18;17:3921-3948. doi: 10.2147/JIR.S466821. eCollection 2024. J Inflamm Res. 2024. PMID: 38911990 Free PMC article. Review.
References
-
- Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England journal of medicine. 2010;362:329–344. - PubMed
-
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nature reviews. Neuroscience. 2007;8:663–672. - PubMed
-
- Ittner LM, Gotz J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nature reviews. Neuroscience. 2011;12:65–72. - PubMed
-
- Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28:202–208. - PubMed
-
- Zlokovic BV. Neurodegeneration and the neurovascular unit. Nat Med. 2010;16:1370–1371. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
