

Hybrid error correction and de novo assembly of single-molecule sequencing reads
- PMID: 22750884
- PMCID: PMC3707490
- DOI: 10.1038/nbt.2280
Hybrid error correction and de novo assembly of single-molecule sequencing reads
Abstract
Single-molecule sequencing instruments can generate multikilobase sequences with the potential to greatly improve genome and transcriptome assembly. However, the error rates of single-molecule reads are high, which has limited their use thus far to resequencing bacteria. To address this limitation, we introduce a correction algorithm and assembly strategy that uses short, high-fidelity sequences to correct the error in single-molecule sequences. We demonstrate the utility of this approach on reads generated by a PacBio RS instrument from phage, prokaryotic and eukaryotic whole genomes, including the previously unsequenced genome of the parrot Melopsittacus undulatus, as well as for RNA-Seq reads of the corn (Zea mays) transcriptome. Our long-read correction achieves >99.9% base-call accuracy, leading to substantially better assemblies than current sequencing strategies: in the best example, the median contig size was quintupled relative to high-coverage, second-generation assemblies. Greater gains are predicted if read lengths continue to increase, including the prospect of single-contig bacterial chromosome assembly.
Conflict of interest statement
Figures
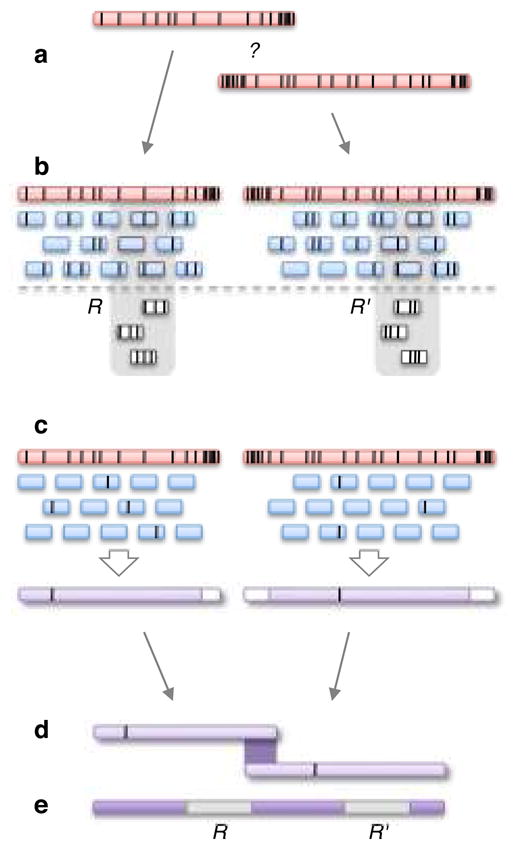
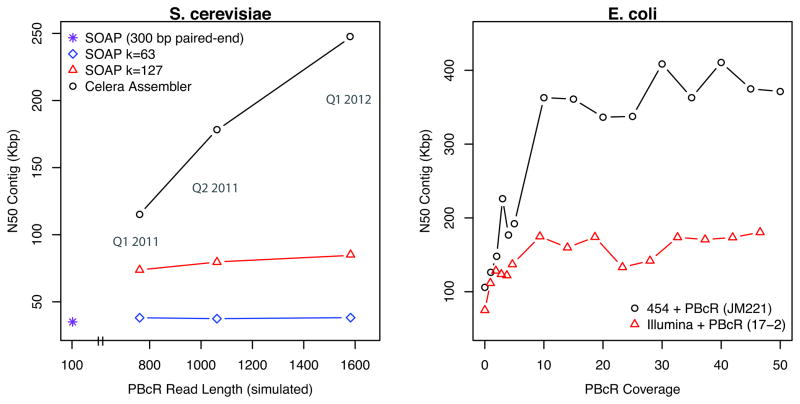
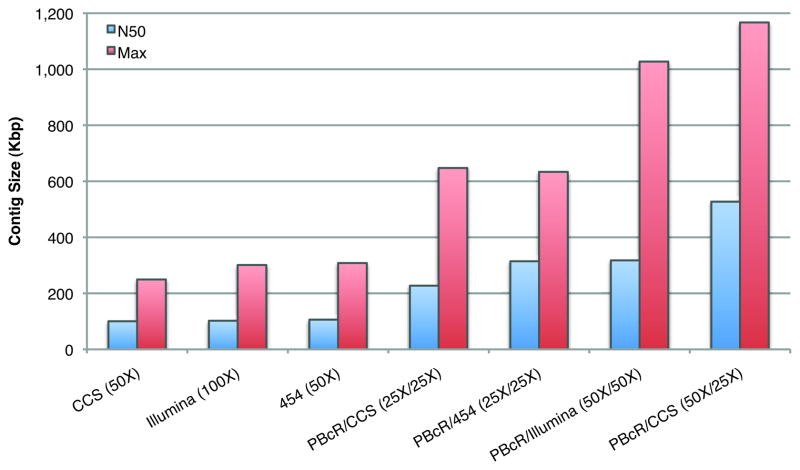
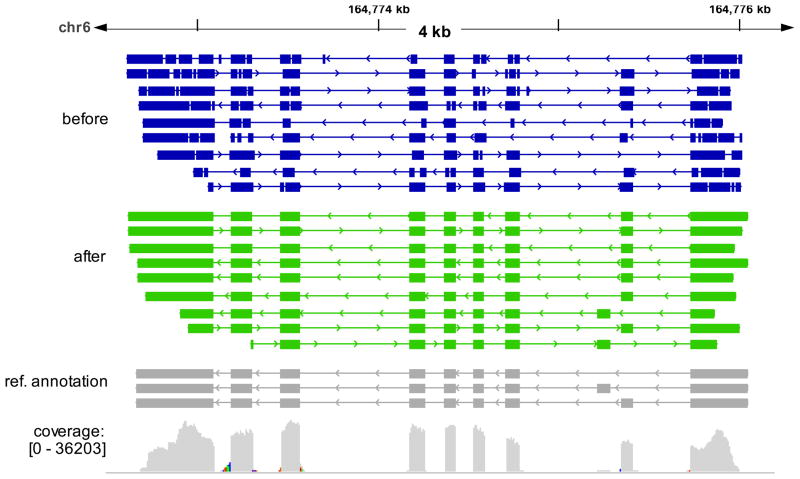
Similar articles
-
A hybrid and scalable error correction algorithm for indel and substitution errors of long reads.BMC Genomics. 2019 Dec 20;20(Suppl 11):948. doi: 10.1186/s12864-019-6286-9. BMC Genomics. 2019. PMID: 31856721 Free PMC article.
-
A de novo Full-Length mRNA Transcriptome Generated From Hybrid-Corrected PacBio Long-Reads Improves the Transcript Annotation and Identifies Thousands of Novel Splice Variants in Atlantic Salmon.Front Genet. 2021 Apr 27;12:656334. doi: 10.3389/fgene.2021.656334. eCollection 2021. Front Genet. 2021. PMID: 33986770 Free PMC article.
-
A comparison of next generation sequencing technologies for transcriptome assembly and utility for RNA-Seq in a non-model bird.PLoS One. 2014 Oct 3;9(10):e108550. doi: 10.1371/journal.pone.0108550. eCollection 2014. PLoS One. 2014. PMID: 25279728 Free PMC article.
-
PacBio Sequencing and Its Applications.Genomics Proteomics Bioinformatics. 2015 Oct;13(5):278-89. doi: 10.1016/j.gpb.2015.08.002. Epub 2015 Nov 2. Genomics Proteomics Bioinformatics. 2015. PMID: 26542840 Free PMC article. Review.
-
De novo assembly of short sequence reads.Brief Bioinform. 2010 Sep;11(5):457-72. doi: 10.1093/bib/bbq020. Epub 2010 Aug 19. Brief Bioinform. 2010. PMID: 20724458 Review.
Cited by
-
Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology.PLoS One. 2012;7(11):e47768. doi: 10.1371/journal.pone.0047768. Epub 2012 Nov 21. PLoS One. 2012. PMID: 23185243 Free PMC article.
-
Cancer whole-genome sequencing: present and future.Oncogene. 2015 Dec 3;34(49):5943-50. doi: 10.1038/onc.2015.90. Epub 2015 Mar 30. Oncogene. 2015. PMID: 25823020 Review.
-
Construction of Pseudomolecules for the Chinese Chestnut (Castanea mollissima) Genome.G3 (Bethesda). 2020 Oct 5;10(10):3565-3574. doi: 10.1534/g3.120.401532. G3 (Bethesda). 2020. PMID: 32847817 Free PMC article.
-
Assembly and diploid architecture of an individual human genome via single-molecule technologies.Nat Methods. 2015 Aug;12(8):780-6. doi: 10.1038/nmeth.3454. Epub 2015 Jun 29. Nat Methods. 2015. PMID: 26121404 Free PMC article.
-
GAML: genome assembly by maximum likelihood.Algorithms Mol Biol. 2015 Jun 3;10:18. doi: 10.1186/s13015-015-0052-6. eCollection 2015. Algorithms Mol Biol. 2015. PMID: 26042154 Free PMC article.
References
-
- Bentley D. Whole-genome re-sequencing. Current Opinion in Genetics & Development. 2006;16:545–552. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
