

AdapterRemoval: easy cleaning of next-generation sequencing reads
- PMID: 22748135
- PMCID: PMC3532080
- DOI: 10.1186/1756-0500-5-337
AdapterRemoval: easy cleaning of next-generation sequencing reads
Abstract
Background: With the advent of next-generation sequencing there is an increased demand for tools to pre-process and handle the vast amounts of data generated. One recurring problem is adapter contamination in the reads, i.e. the partial or complete sequencing of adapter sequences. These adapter sequences have to be removed as they can hinder correct mapping of the reads and influence SNP calling and other downstream analyses.
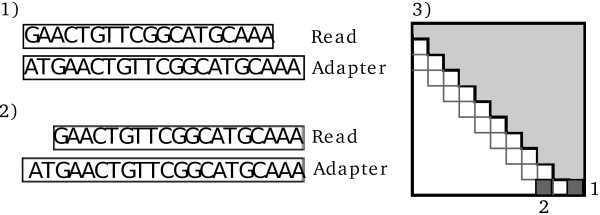
Findings: We present a tool called AdapterRemoval which is able to pre-process both single and paired-end data. The program locates and removes adapter residues from the reads, it is able to combine paired reads if they overlap, and it can optionally trim low-quality nucleotides. Furthermore, it can look for adapter sequence in both the 5' and 3' ends of the reads. This is a flexible tool that can be tuned to accommodate different experimental settings and sequencing platforms producing FASTQ files. AdapterRemoval is shown to be good at trimming adapters from both single-end and paired-end data.
Conclusions: AdapterRemoval is a comprehensive tool for analyzing next-generation sequencing data. It exhibits good performance both in terms of sensitivity and specificity. AdapterRemoval has already been used in various large projects and it is possible to extend it further to accommodate application-specific biases in the data.
Figures

Similar articles
-
AdapterRemoval v2: rapid adapter trimming, identification, and read merging.BMC Res Notes. 2016 Feb 12;9:88. doi: 10.1186/s13104-016-1900-2. BMC Res Notes. 2016. PMID: 26868221 Free PMC article.
-
Sequence-matching adapter trimmers generate consistent quality and assembly metrics for Illumina sequencing of RNA viruses.BMC Res Notes. 2024 Oct 14;17(1):308. doi: 10.1186/s13104-024-06951-0. BMC Res Notes. 2024. PMID: 39402647 Free PMC article.
-
Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads.BMC Bioinformatics. 2014 Jun 12;15:182. doi: 10.1186/1471-2105-15-182. BMC Bioinformatics. 2014. PMID: 24925680 Free PMC article.
-
Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies.Genomics. 2011 Aug;98(2):152-3. doi: 10.1016/j.ygeno.2011.05.009. Epub 2011 May 30. Genomics. 2011. PMID: 21651976
-
MiSeq: A Next Generation Sequencing Platform for Genomic Analysis.Methods Mol Biol. 2018;1706:223-232. doi: 10.1007/978-1-4939-7471-9_12. Methods Mol Biol. 2018. PMID: 29423801 Review.
Cited by
-
In vivo treatment with a non-aromatizable androgen rapidly alters the ovarian transcriptome of previtellogenic secondary growth coho salmon (Onchorhynchus kisutch).PLoS One. 2024 Oct 9;19(10):e0311628. doi: 10.1371/journal.pone.0311628. eCollection 2024. PLoS One. 2024. PMID: 39383164 Free PMC article.
-
Female sex bias in Iberian megalithic societies through bioarchaeology, aDNA and proteomics.Sci Rep. 2024 Sep 23;14(1):21818. doi: 10.1038/s41598-024-72148-x. Sci Rep. 2024. PMID: 39313501 Free PMC article.
-
Long genetic and social isolation in Neanderthals before their extinction.Cell Genom. 2024 Sep 11;4(9):100593. doi: 10.1016/j.xgen.2024.100593. Cell Genom. 2024. PMID: 39265525 Free PMC article.
-
Ancient Rapanui genomes reveal resilience and pre-European contact with the Americas.Nature. 2024 Sep;633(8029):389-397. doi: 10.1038/s41586-024-07881-4. Epub 2024 Sep 11. Nature. 2024. PMID: 39261618 Free PMC article.
-
Whole-Genome Resequencing Analysis of the Camelus bactrianus (Bactrian Camel) Genome Identifies Mutations and Genes Affecting Milk Production Traits.Int J Mol Sci. 2024 Jul 17;25(14):7836. doi: 10.3390/ijms25147836. Int J Mol Sci. 2024. PMID: 39063078 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
