

doi: 10.1038/nmeth.2066.
Metagenomic microbial community profiling using unique clade-specific marker genes
Affiliations
- PMID: 22688413
- PMCID: PMC3443552
- DOI: 10.1038/nmeth.2066
Item in Clipboard
Metagenomic microbial community profiling using unique clade-specific marker genes
Nat Methods.
.
Abstract
Metagenomic shotgun sequencing data can identify microbes populating a microbial community and their proportions, but existing taxonomic profiling methods are inefficient for increasingly large data sets. We present an approach that uses clade-specific marker genes to unambiguously assign reads to microbial clades more accurately and >50× faster than current approaches. We validated our metagenomic phylogenetic analysis tool, MetaPhlAn, on terabases of short reads and provide the largest metagenomic profiling to date of the human gut. It can be accessed at http://huttenhower.sph.harvard.edu/metaphlan/.
Figures
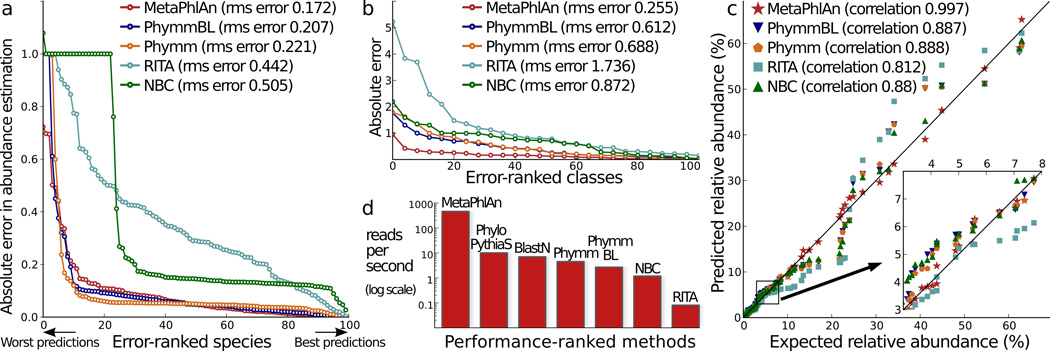
We used 10 total synthetic metagenomes to compare MetaPhlAn to PhymmBL, BLAST, theRITA pipeline, and NBC. a–b) Absolute and root mean squared (rms) errors with respect to 100 total organisms in one synthetic metagenome at the species (a) and class (b) level c) Correlations of inferred and true species abundances on 8 non-evenly distributed synthetic metagenomes;. d) Reads-per-second rates for the tested methods on single CPUs.

MetaPhlAn species and genus abundances and 16S phylotype abundances for 51 healthy vaginal microbiomes from the Human Microbiome Project. Samples were naively grouped by assigning each based on its dominant (>50%) Lactobacillus species or by the absence (<2%) of any Lactobacillus. For each cluster (named from I to V) we report averages across samples for all genera and species as inferred by MetaPhlAn and, for genera, as estimated by the combination of mothur and the RDP classifier (see Methods) applied to 16S rRNA gene sequences from the same specimen.

a) A taxonomic cladogram reporting all clades present in one or both cohorts (≥0.5% abundance in ≥1 sample). Circle size is proportional to the log of average abundance; color represents relative enrichment of the most abundant taxa (≥1% average in ≥1 cohort) between the HMP (139) and MetaHIT (85, healthy only) populations. b–c) Genus- (b) and species-level (c) taxonomic profiles of the most abundant clades hierarchically clustered (average linkage) with the Bray-Curtis similarity reveal sets of samples with similar microbial community compositions. With the exception of the cluster dominated by genus Bacteroides (B. vulgatus and B. ovatus in particular), samples from both studies are present in all groups, confirming substantial consistency of the gut microbiota characterized by independent and geographically distant western-diet asymptomatic cohorts. Only species and genera with at least 7.5% abundances at the 95th percentile of their distribution are reported.
Comment in
-
High-speed microbial community profiling.Nat Methods. 2012 Jun 10;9(8):793-4. doi: 10.1038/nmeth.2080. Nat Methods. 2012. PMID: 22688412 No abstract available.
Similar articles
-
Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences.BMC Genomics. 2011;12 Suppl 2(Suppl 2):S4. doi: 10.1186/1471-2164-12-S2-S4. Epub 2011 Jul 27. BMC Genomics. 2011. PMID: 21989143 Free PMC article.
-
Assembly-free metagenomic analysis reveals new metabolic capabilities in surface ocean bacterioplankton.Environ Microbiol Rep. 2013 Oct;5(5):686-96. doi: 10.1111/1758-2229.12068. Epub 2013 May 16. Environ Microbiol Rep. 2013. PMID: 24115619
-
Selection of marker genes for genetic barcoding of microorganisms and binning of metagenomic reads by Barcoder software tools.BMC Bioinformatics. 2018 Aug 30;19(1):309. doi: 10.1186/s12859-018-2320-1. BMC Bioinformatics. 2018. PMID: 30165813 Free PMC article.
-
Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses.Microb Genom. 2020 Aug;6(8):mgen000409. doi: 10.1099/mgen.0.000409. Epub 2020 Jul 24. Microb Genom. 2020. PMID: 32706331 Free PMC article. Review.
-
Application of computational approaches to analyze metagenomic data.J Microbiol. 2021 Mar;59(3):233-241. doi: 10.1007/s12275-021-0632-8. Epub 2021 Feb 10. J Microbiol. 2021. PMID: 33565054 Review.
Cited by
-
Optimized DNA extraction and metagenomic sequencing of airborne microbial communities.Nat Protoc. 2015 May;10(5):768-79. doi: 10.1038/nprot.2015.046. Epub 2015 Apr 23. Nat Protoc. 2015. PMID: 25906115 Free PMC article.
-
Historic methicillin-resistant Staphylococcus aureus: expanding current knowledge using molecular epidemiological characterization of a Swiss legacy collection.Genome Med. 2024 Feb 5;16(1):23. doi: 10.1186/s13073-024-01292-w. Genome Med. 2024. PMID: 38317199 Free PMC article.
-
MTSv: rapid alignment-based taxonomic classification and high-confidence metagenomic analysis.PeerJ. 2022 Nov 8;10:e14292. doi: 10.7717/peerj.14292. eCollection 2022. PeerJ. 2022. PMID: 36389404 Free PMC article.
-
Filtration and Normalization of Sequencing Read Data in Whole-Metagenome Shotgun Samples.PLoS One. 2016 Oct 19;11(10):e0165015. doi: 10.1371/journal.pone.0165015. eCollection 2016. PLoS One. 2016. PMID: 27760173 Free PMC article.
-
DOE JGI Metagenome Workflow.mSystems. 2021 May 18;6(3):e00804-20. doi: 10.1128/mSystems.00804-20. mSystems. 2021. PMID: 34006627 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
