

Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration
- PMID: 22647602
- PMCID: PMC3386132
- DOI: 10.1073/pnas.1112368109
Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration
Abstract
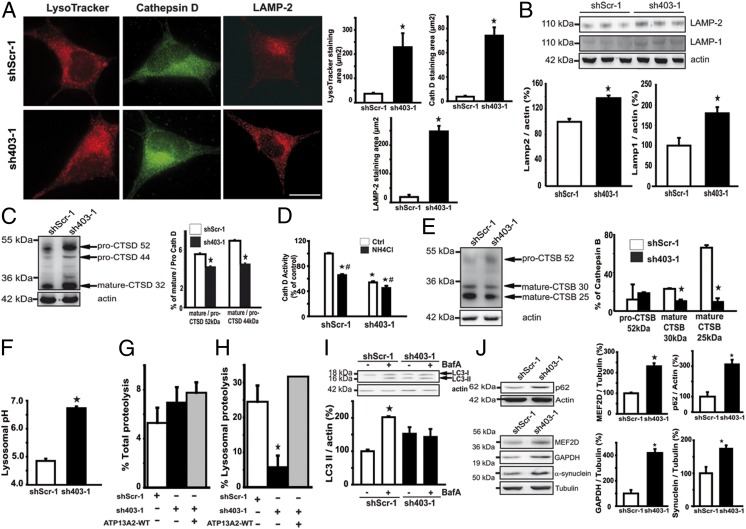
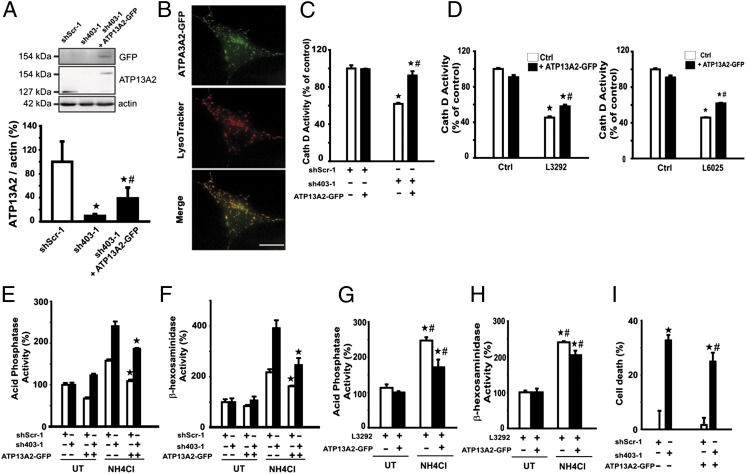
Parkinson disease (PD) is a progressive neurodegenerative disorder pathologically characterized by the loss of dopaminergic neurons from the substantia nigra pars compacta and the presence, in affected brain regions, of protein inclusions named Lewy bodies (LBs). The ATP13A2 gene (locus PARK9) encodes the protein ATP13A2, a lysosomal type 5 P-type ATPase that is linked to autosomal recessive familial parkinsonism. The physiological function of ATP13A2, and hence its role in PD, remains to be elucidated. Here, we show that PD-linked mutations in ATP13A2 lead to several lysosomal alterations in ATP13A2 PD patient-derived fibroblasts, including impaired lysosomal acidification, decreased proteolytic processing of lysosomal enzymes, reduced degradation of lysosomal substrates, and diminished lysosomal-mediated clearance of autophagosomes. Similar alterations are observed in stable ATP13A2-knockdown dopaminergic cell lines, which are associated with cell death. Restoration of ATP13A2 levels in ATP13A2-mutant/depleted cells restores lysosomal function and attenuates cell death. Relevant to PD, ATP13A2 levels are decreased in dopaminergic nigral neurons from patients with PD, in which ATP13A2 mostly accumulates within Lewy bodies. Our results unravel an instrumental role of ATP13A2 deficiency on lysosomal function and cell viability and demonstrate the feasibility and therapeutic potential of modulating ATP13A2 levels in the context of PD.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Comment in
-
New insights into lysosomal dysfunction in Parkinson’s disease: an emerging role for ATP13A2.Mov Disord. 2012 Aug;27(9):1092. doi: 10.1002/mds.25118. Mov Disord. 2012. PMID: 23035259 No abstract available.
Similar articles
-
Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove.Autophagy. 2012 Sep;8(9):1389-91. doi: 10.4161/auto.21011. Epub 2012 Aug 13. Autophagy. 2012. PMID: 22885599 Free PMC article.
-
Neuropathology in an α-synuclein preformed fibril mouse model occurs independent of the Parkinson's disease-linked lysosomal ATP13A2 protein.Neurobiol Dis. 2024 Nov;202:106701. doi: 10.1016/j.nbd.2024.106701. Epub 2024 Oct 13. Neurobiol Dis. 2024. PMID: 39406291
-
PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity.Hum Mol Genet. 2012 Apr 15;21(8):1725-43. doi: 10.1093/hmg/ddr606. Epub 2011 Dec 20. Hum Mol Genet. 2012. PMID: 22186024 Free PMC article.
-
Lysosomal impairment in Parkinson's disease.Mov Disord. 2013 Jun;28(6):725-32. doi: 10.1002/mds.25462. Epub 2013 Apr 11. Mov Disord. 2013. PMID: 23580333 Free PMC article. Review.
-
Hereditary Parkinsonism-Associated Genetic Variations in PARK9 Locus Lead to Functional Impairment of ATPase Type 13A2.Curr Protein Pept Sci. 2017;18(7):725-732. doi: 10.2174/1389203717666160311121534. Curr Protein Pept Sci. 2017. PMID: 26965689 Review.
Cited by
-
Sestrin2 Protects Dopaminergic Cells against Rotenone Toxicity through AMPK-Dependent Autophagy Activation.Mol Cell Biol. 2015 Aug;35(16):2740-51. doi: 10.1128/MCB.00285-15. Epub 2015 Jun 1. Mol Cell Biol. 2015. PMID: 26031332 Free PMC article.
-
Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson's Disease.Int J Mol Sci. 2023 Dec 26;25(1):360. doi: 10.3390/ijms25010360. Int J Mol Sci. 2023. PMID: 38203531 Free PMC article. Review.
-
Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification.Eur J Neurosci. 2013 Jun;37(12):1949-61. doi: 10.1111/ejn.12169. Eur J Neurosci. 2013. PMID: 23773064 Free PMC article.
-
Amyloid-like aggregating proteins cause lysosomal defects in neurons via gain-of-function toxicity.Life Sci Alliance. 2021 Dec 21;5(3):e202101185. doi: 10.26508/lsa.202101185. Print 2022 Mar. Life Sci Alliance. 2021. PMID: 34933920 Free PMC article.
-
Vesicular Dysfunction and the Pathogenesis of Parkinson's Disease: Clues From Genetic Studies.Front Neurosci. 2020 Jan 8;13:1381. doi: 10.3389/fnins.2019.01381. eCollection 2019. Front Neurosci. 2020. PMID: 31969802 Free PMC article. Review.
References
-
- Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and models. Neuron. 2003;39:889–909. - PubMed
-
- Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. 1994;89:347–352. - PubMed
-
- Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. - PubMed
-
- Di Fonzo A, et al. Italian Parkinson Genetics Network ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–1562. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
