

Physical tethering and volume exclusion determine higher-order genome organization in budding yeast
- PMID: 22619363
- PMCID: PMC3396370
- DOI: 10.1101/gr.129437.111
Physical tethering and volume exclusion determine higher-order genome organization in budding yeast
Abstract
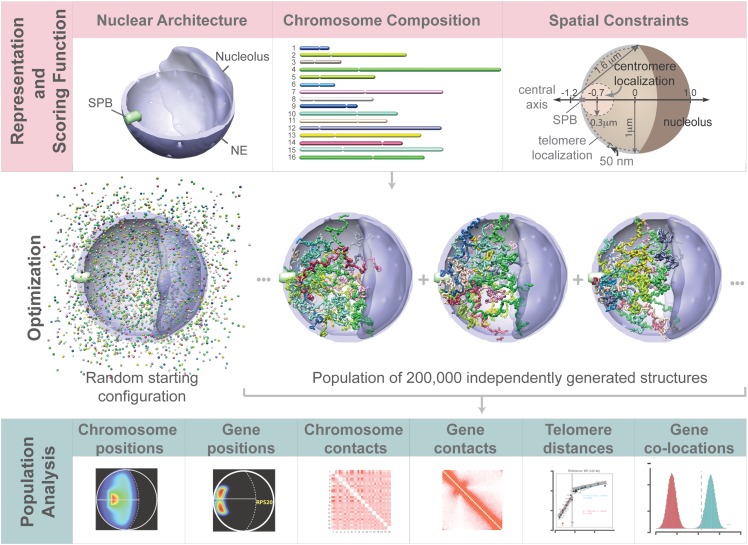
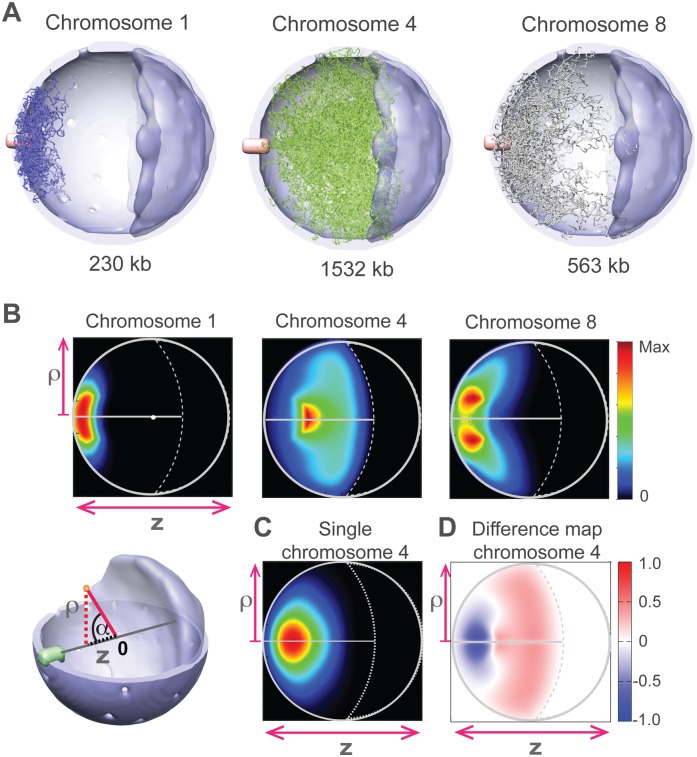
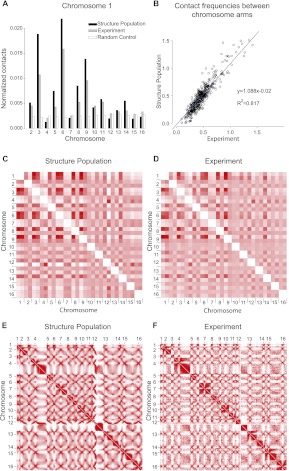
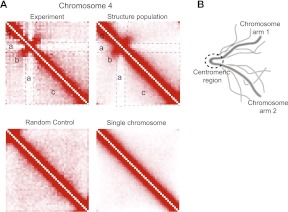
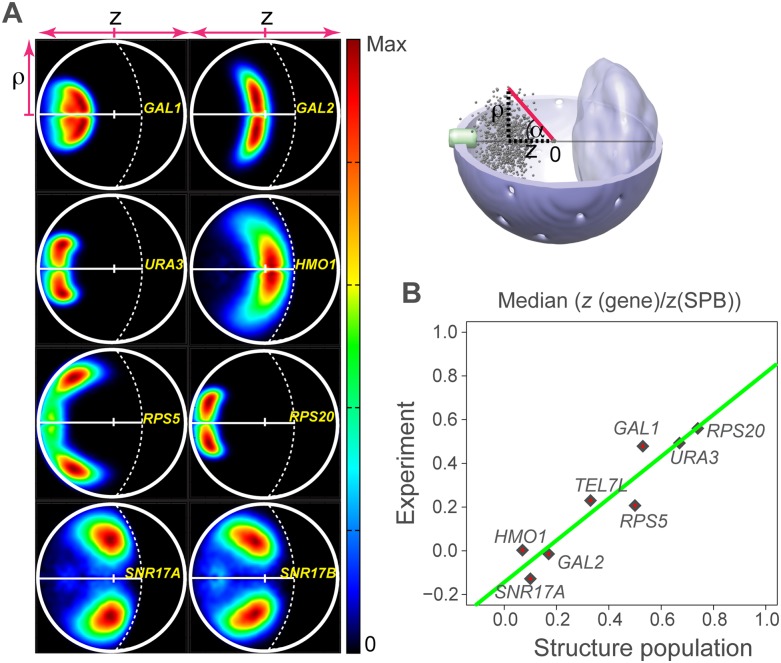
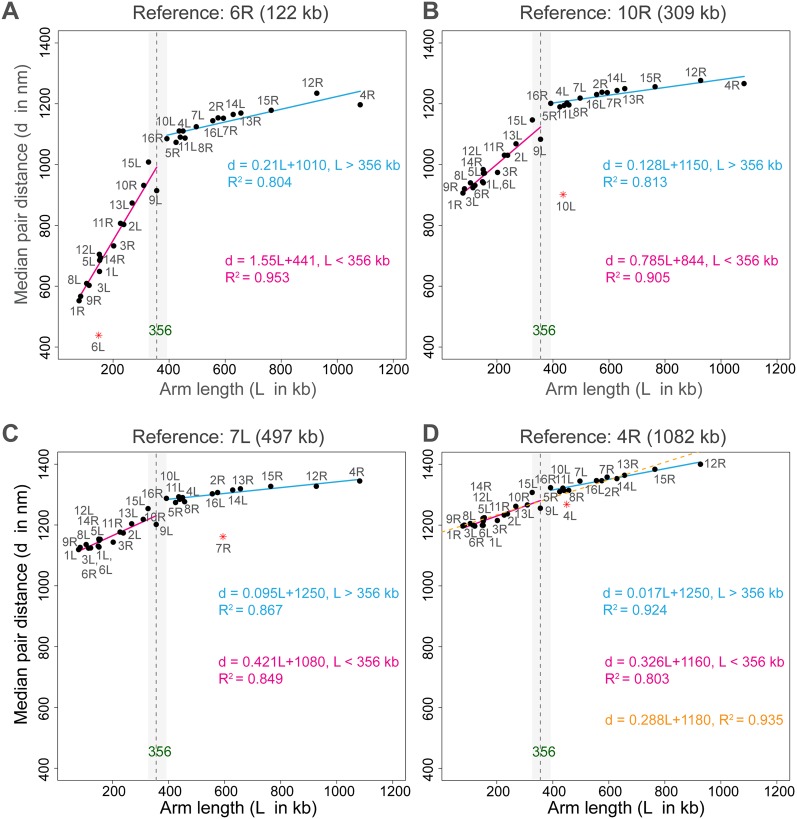
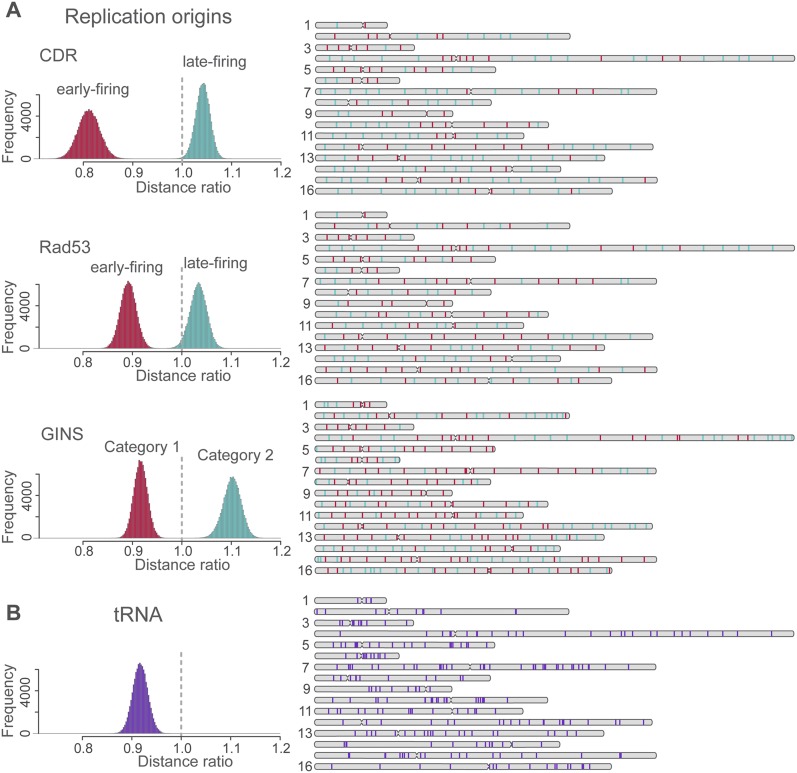
In this paper we show that tethering of heterochromatic regions to nuclear landmarks and random encounters of chromosomes in the confined nuclear volume are sufficient to explain the higher-order organization of the budding yeast genome. We have quantitatively characterized the contact patterns and nuclear territories that emerge when chromosomes are allowed to behave as constrained but otherwise randomly configured flexible polymer chains in the nucleus. Remarkably, this constrained random encounter model explains in a statistical manner the experimental hallmarks of the S. cerevisiae genome organization, including (1) the folding patterns of individual chromosomes; (2) the highly enriched interactions between specific chromatin regions and chromosomes; (3) the emergence, shape, and position of gene territories; (4) the mean distances between pairs of telomeres; and (5) even the co-location of functionally related gene loci, including early replication start sites and tRNA genes. Therefore, most aspects of the yeast genome organization can be explained without calling on biochemically mediated chromatin interactions. Such interactions may modulate the pre-existing propensity for co-localization but seem not to be the cause for the observed higher-order organization. The fact that geometrical constraints alone yield a highly organized genome structure, on which different functional elements are specifically distributed, has strong implications for the folding principles of the genome and the evolution of its function.
Figures
Similar articles
-
A three-dimensional model of the yeast genome.Nature. 2010 May 20;465(7296):363-7. doi: 10.1038/nature08973. Epub 2010 May 2. Nature. 2010. PMID: 20436457 Free PMC article.
-
Comparative 3D genome structure analysis of the fission and the budding yeast.PLoS One. 2015 Mar 23;10(3):e0119672. doi: 10.1371/journal.pone.0119672. eCollection 2015. PLoS One. 2015. PMID: 25799503 Free PMC article.
-
Chromosome positioning and the clustering of functionally related loci in yeast is driven by chromosomal interactions.Nucleus. 2012 Jul 1;3(4):370-83. doi: 10.4161/nucl.20971. Epub 2012 Jun 12. Nucleus. 2012. PMID: 22688649
-
Nuclear organization and chromatin dynamics in yeast: biophysical models or biologically driven interactions?Biochim Biophys Acta. 2012 Jun;1819(6):468-81. doi: 10.1016/j.bbagrm.2011.12.010. Epub 2012 Jan 5. Biochim Biophys Acta. 2012. PMID: 22245105 Review.
-
Principles of chromosomal organization: lessons from yeast.J Cell Biol. 2011 Mar 7;192(5):723-33. doi: 10.1083/jcb.201010058. J Cell Biol. 2011. PMID: 21383075 Free PMC article. Review.
Cited by
-
Impact of Chromosome Fusions on 3D Genome Organization and Gene Expression in Budding Yeast.Genetics. 2020 Mar;214(3):651-667. doi: 10.1534/genetics.119.302978. Epub 2020 Jan 6. Genetics. 2020. PMID: 31907200 Free PMC article.
-
Improved accuracy assessment for 3D genome reconstructions.BMC Bioinformatics. 2018 May 30;19(1):196. doi: 10.1186/s12859-018-2214-2. BMC Bioinformatics. 2018. PMID: 29848293 Free PMC article.
-
Unsupervised embedding of single-cell Hi-C data.Bioinformatics. 2018 Jul 1;34(13):i96-i104. doi: 10.1093/bioinformatics/bty285. Bioinformatics. 2018. PMID: 29950005 Free PMC article.
-
A statistical approach for inferring the 3D structure of the genome.Bioinformatics. 2014 Jun 15;30(12):i26-33. doi: 10.1093/bioinformatics/btu268. Bioinformatics. 2014. PMID: 24931992 Free PMC article.
-
The 3D Genome as Moderator of Chromosomal Communication.Cell. 2016 Mar 10;164(6):1110-1121. doi: 10.1016/j.cell.2016.02.007. Cell. 2016. PMID: 26967279 Free PMC article. Review.
References
-
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. 2007a. Determining the architectures of macromolecular assemblies. Nature 450: 683–694 - PubMed
-
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. 2007b. The molecular architecture of the nuclear pore complex. Nature 450: 695–701 - PubMed
-
- Alber F, Forster F, Korkin D, Topf M, Sali A 2008. Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem 77: 443–477 - PubMed
-
- Berger AB, Cabal GG, Fabre E, Duong T, Buc H, Nehrbass U, Olivo-Marin JC, Gadal O, Zimmer C 2008. High-resolution statistical mapping reveals gene territories in live yeast. Nat Methods 5: 1031–1037 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases