

Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies
- PMID: 22613949
- PMCID: PMC3411175
- DOI: 10.1038/cr.2012.82
Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies
Abstract
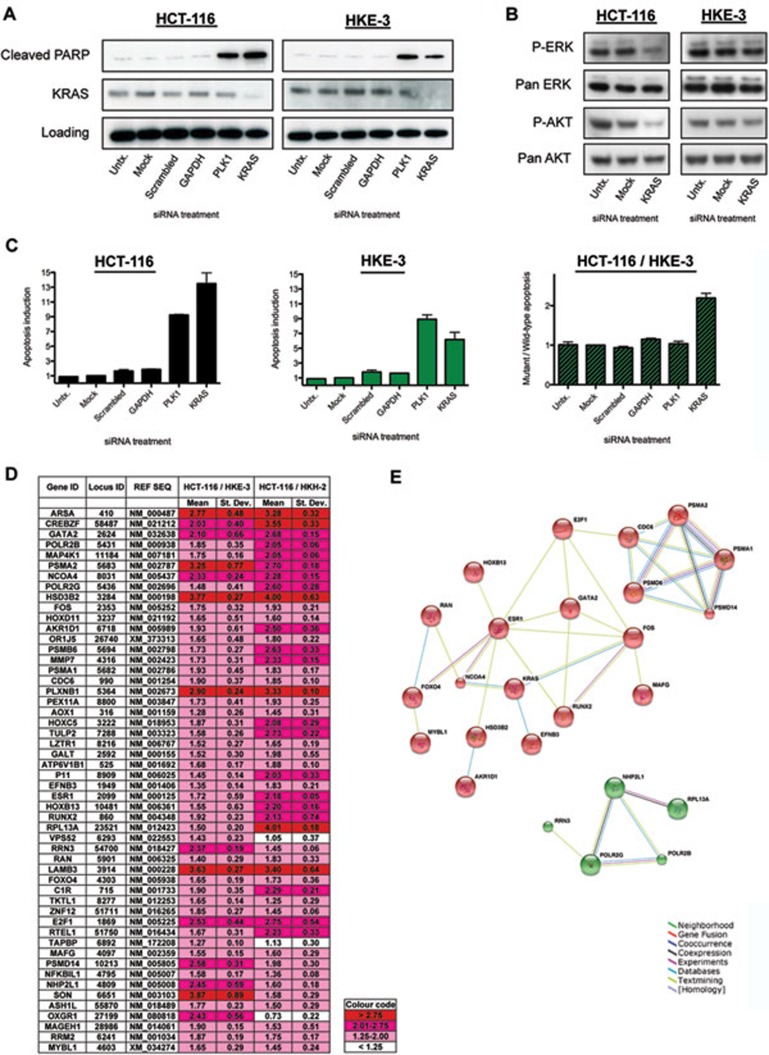
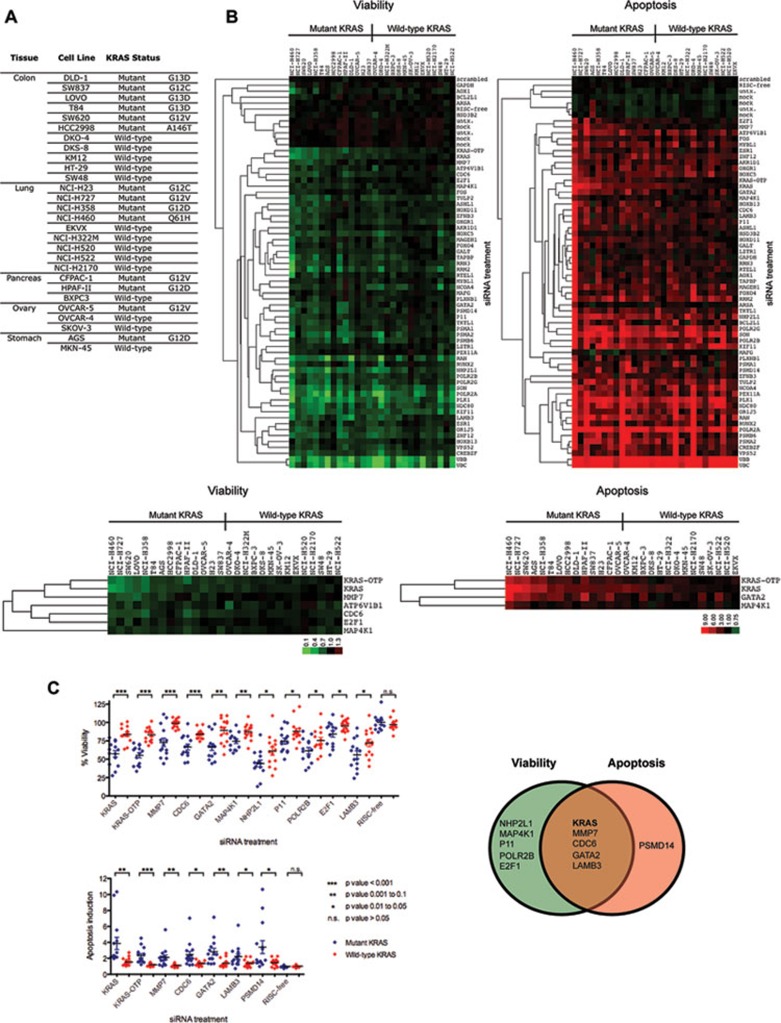
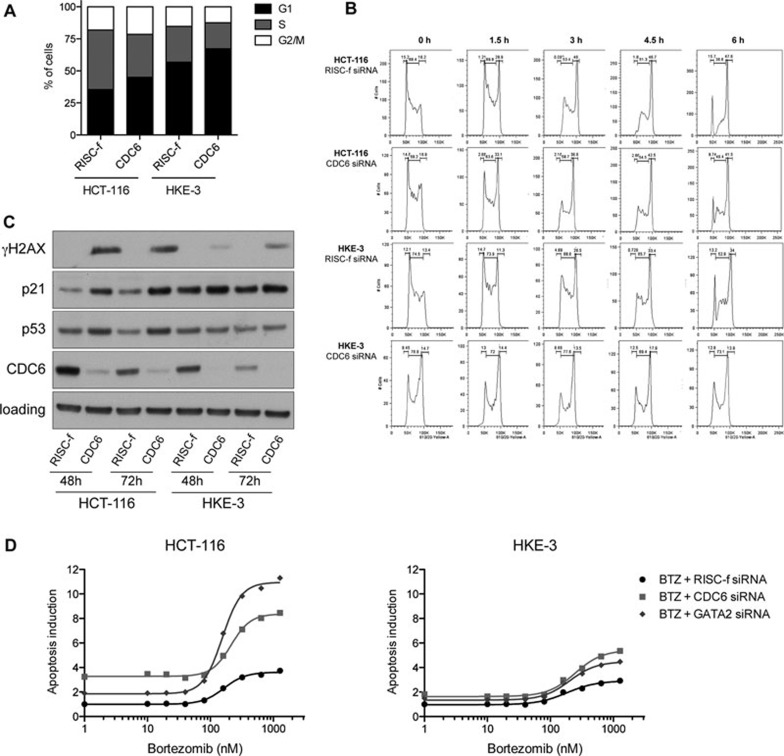
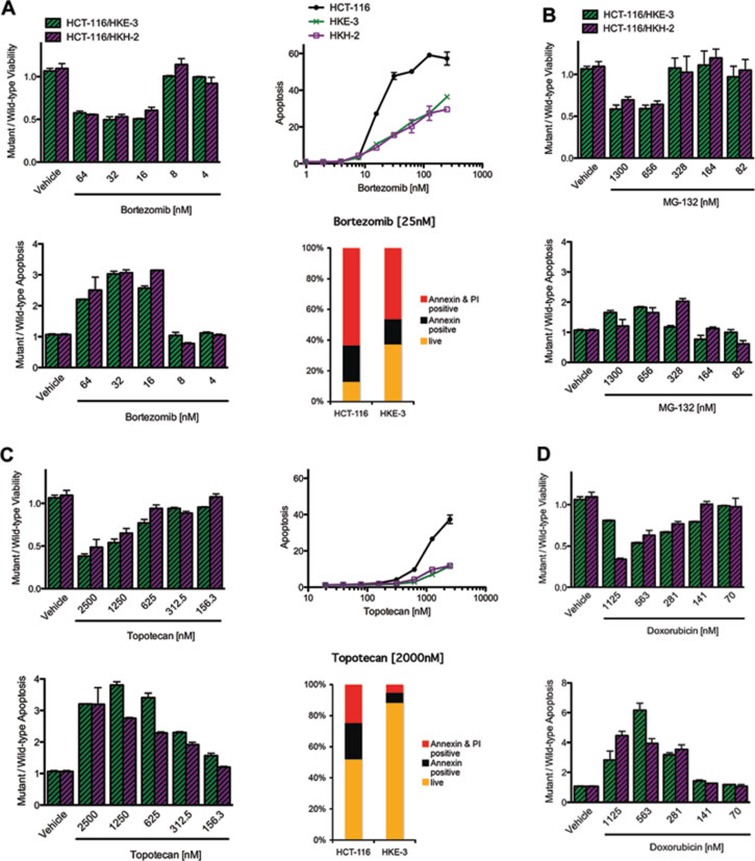
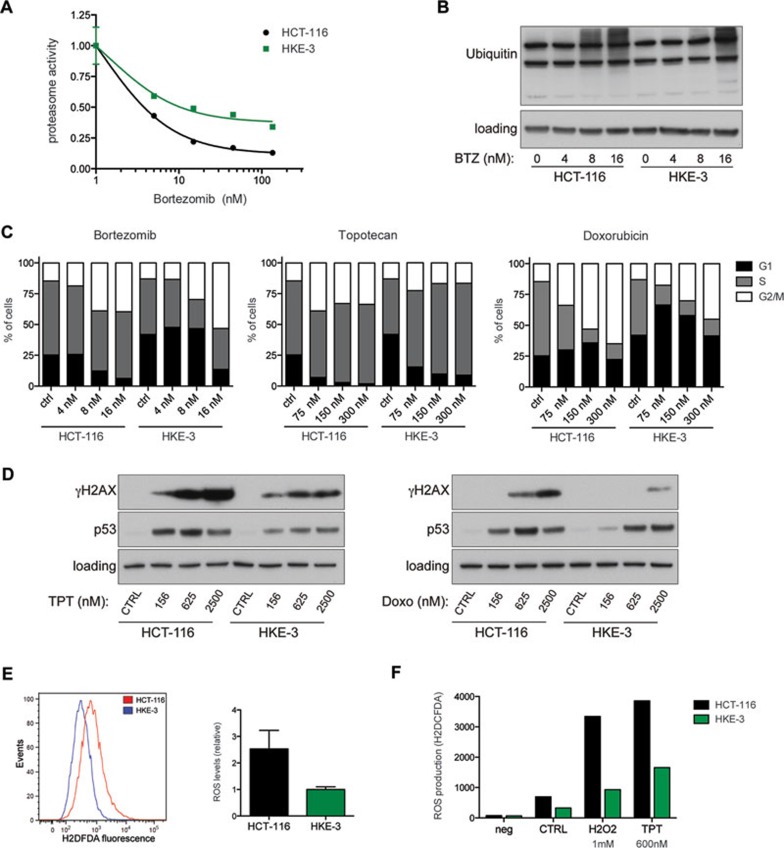
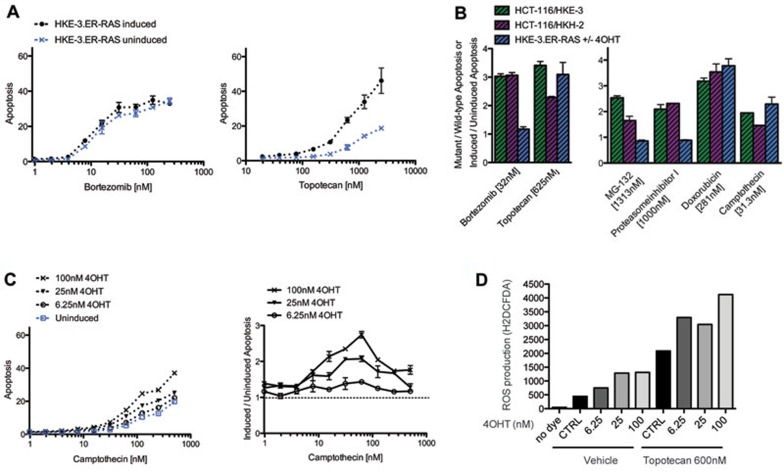
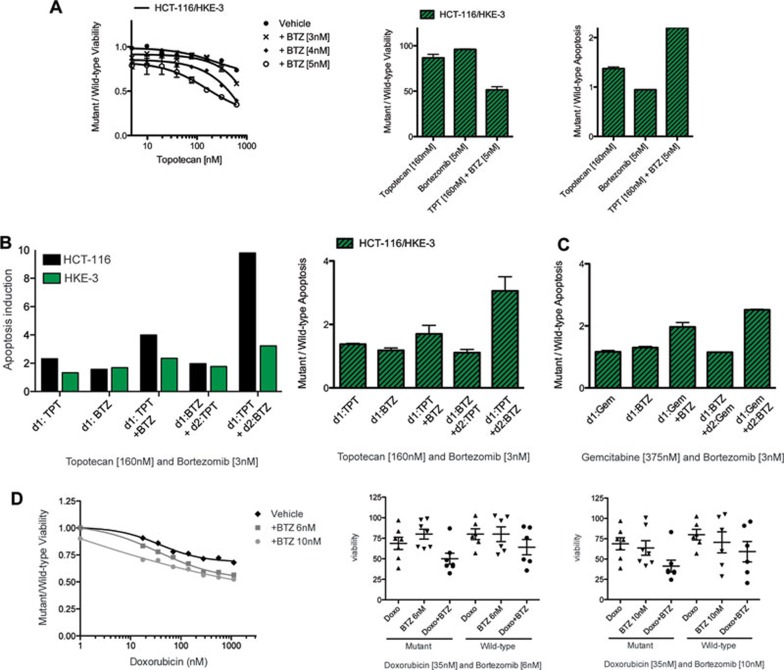
Oncogenic mutations in RAS genes are very common in human cancer, resulting in cells with well-characterized selective advantages, but also less well-understood vulnerabilities. We have carried out a large-scale loss-of-function screen to identify genes that are required by KRAS-transformed colon cancer cells, but not by derivatives lacking this oncogene. Top-scoring genes were then tested in a larger panel of KRAS mutant and wild-type cancer cells. Cancer cells expressing oncogenic KRAS were found to be highly dependent on the transcription factor GATA2 and the DNA replication initiation regulator CDC6. Extending this analysis using a collection of drugs with known targets, we found that cancer cells with mutant KRAS showed selective addiction to proteasome function, as well as synthetic lethality with topoisomerase inhibition. Combination targeting of these functions caused improved killing of KRAS mutant cells relative to wild-type cells. These observations suggest novel targets and new ways of combining existing therapies for optimal effect in RAS mutant cancers, which are traditionally seen as being highly refractory to therapy.
Figures
Similar articles
-
c-Met targeting enhances the effect of irradiation and chemical agents against malignant colon cells harboring a KRAS mutation.PLoS One. 2014 Nov 26;9(11):e113186. doi: 10.1371/journal.pone.0113186. eCollection 2014. PLoS One. 2014. PMID: 25427200 Free PMC article.
-
A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene.Cell. 2009 May 29;137(5):835-48. doi: 10.1016/j.cell.2009.05.006. Cell. 2009. PMID: 19490893 Free PMC article.
-
The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer.Cell. 2012 Apr 27;149(3):642-55. doi: 10.1016/j.cell.2012.02.059. Cell. 2012. PMID: 22541434
-
The genetics and biology of KRAS in lung cancer.Chin J Cancer. 2013 Feb;32(2):63-70. doi: 10.5732/cjc.012.10098. Epub 2012 Jul 2. Chin J Cancer. 2013. PMID: 22776234 Free PMC article. Review.
-
Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy?J Clin Oncol. 2010 Nov 1;28(31):4769-77. doi: 10.1200/JCO.2009.27.4365. Epub 2010 Oct 4. J Clin Oncol. 2010. PMID: 20921461 Review.
Cited by
-
Development of synthetic lethality in cancer: molecular and cellular classification.Signal Transduct Target Ther. 2020 Oct 19;5(1):241. doi: 10.1038/s41392-020-00358-6. Signal Transduct Target Ther. 2020. PMID: 33077733 Free PMC article. Review.
-
SL-BioDP: Multi-Cancer Interactive Tool for Prediction of Synthetic Lethality and Response to Cancer Treatment.Cancers (Basel). 2019 Oct 29;11(11):1682. doi: 10.3390/cancers11111682. Cancers (Basel). 2019. PMID: 31671773 Free PMC article.
-
Efficient Correction of Oncogenic KRAS and TP53 Mutations through CRISPR Base Editing.Cancer Res. 2022 Sep 2;82(17):3002-3015. doi: 10.1158/0008-5472.CAN-21-2519. Cancer Res. 2022. PMID: 35802645 Free PMC article.
-
Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA).Blood. 2020 Jul 2;136(1):24-35. doi: 10.1182/blood.2019000937. Blood. 2020. PMID: 32430494 Free PMC article. Review.
-
Pharmacotherapeutic Management of Pancreatic Ductal Adenocarcinoma: Current and Emerging Concepts.Drugs Aging. 2017 May;34(5):331-357. doi: 10.1007/s40266-017-0453-y. Drugs Aging. 2017. PMID: 28349415 Review.
References
-
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. - PubMed
-
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. - PubMed
-
- Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. - PubMed
-
- Weir B, Zhao X, Meyerson M. Somatic alterations in the human cancer genome. Cancer Cell. 2004;6:433–438. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
