

From genome-wide association studies to disease mechanisms: celiac disease as a model for autoimmune diseases
- PMID: 22580835
- PMCID: PMC3410018
- DOI: 10.1007/s00281-012-0312-1
From genome-wide association studies to disease mechanisms: celiac disease as a model for autoimmune diseases
Abstract
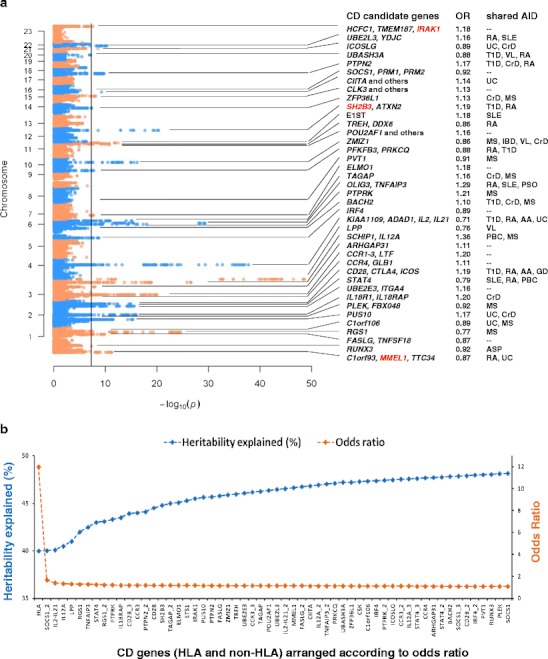
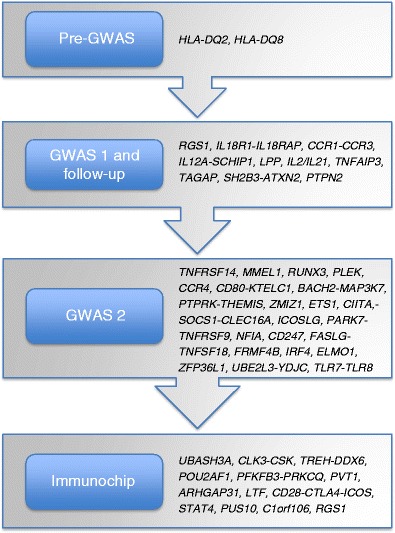
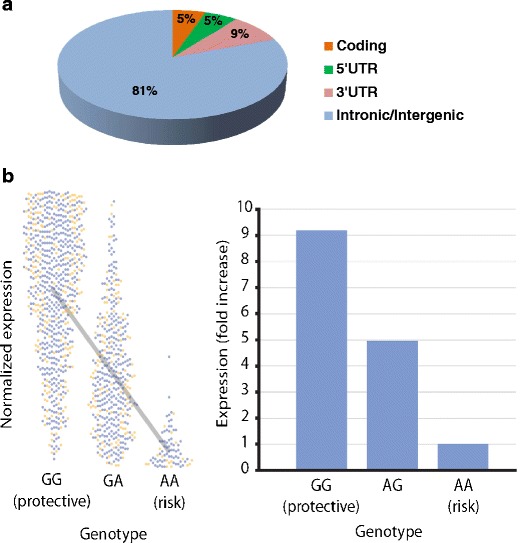
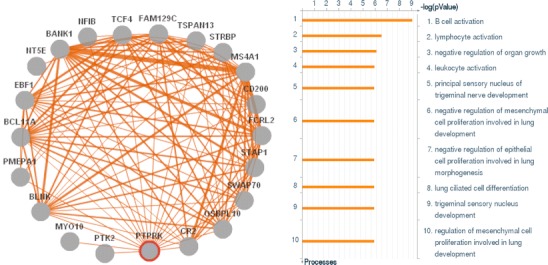
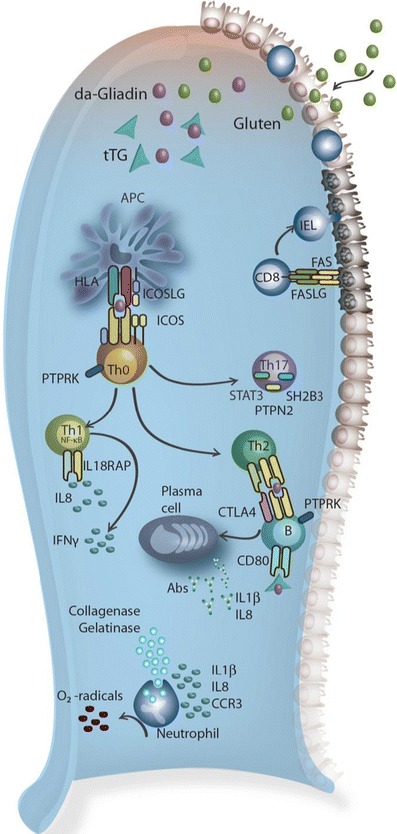
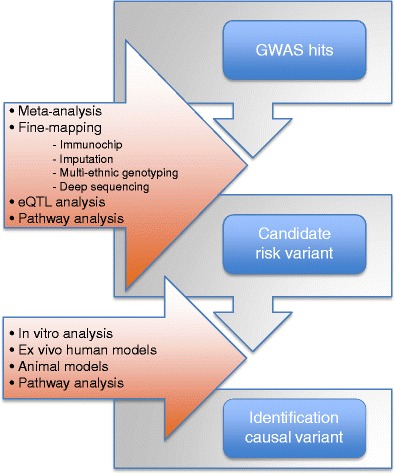
Celiac disease is characterized by a chronic inflammatory reaction in the intestine and is triggered by gluten, a constituent derived from grains which is present in the common daily diet in the Western world. Despite decades of research, the mechanisms behind celiac disease etiology are still not fully understood, although it is clear that both genetic and environmental factors are involved. To improve the understanding of the disease, the genetic component has been extensively studied by genome-wide association studies. These have uncovered a wealth of information that still needs further investigation to clarify its importance. In this review, we summarize and discuss the results of the genetic studies in celiac disease, focusing on the "non-HLA" genes. We also present novel approaches to identifying the causal variants in complex susceptibility loci and disease mechanisms.
Figures
Similar articles
-
A possible mechanism behind autoimmune disorders discovered by genome-wide linkage and association analysis in celiac disease.PLoS One. 2013 Aug 2;8(8):e70174. doi: 10.1371/journal.pone.0070174. Print 2013. PLoS One. 2013. PMID: 23936387 Free PMC article.
-
Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non-coding RNAs.J Autoimmun. 2016 Apr;68:62-74. doi: 10.1016/j.jaut.2016.01.002. Epub 2016 Feb 18. J Autoimmun. 2016. PMID: 26898941 Free PMC article.
-
Celiac disease: moving from genetic associations to causal variants.Clin Genet. 2011 Sep;80(3):203-313. doi: 10.1111/j.1399-0004.2011.01707.x. Epub 2011 Jun 13. Clin Genet. 2011. PMID: 21595655 Review.
-
Celiac disease and endocrine autoimmunity - the genetic link.Autoimmun Rev. 2018 Dec;17(12):1169-1175. doi: 10.1016/j.autrev.2018.05.013. Epub 2018 Oct 12. Autoimmun Rev. 2018. PMID: 30316996 Review.
-
Sex bias in celiac disease: XWAS and monocyte eQTLs in women identify TMEM187 as a functional candidate gene.Biol Sex Differ. 2023 Dec 11;14(1):86. doi: 10.1186/s13293-023-00572-1. Biol Sex Differ. 2023. PMID: 38072919 Free PMC article.
Cited by
-
Review article: Becoming and being coeliac-special considerations for childhood, adolescence and beyond.Aliment Pharmacol Ther. 2022 Jul;56 Suppl 1(Suppl 1):S73-S85. doi: 10.1111/apt.16851. Aliment Pharmacol Ther. 2022. PMID: 35815825 Free PMC article. Review.
-
Immunochip SNP array identifies novel genetic variants conferring susceptibility to candidaemia.Nat Commun. 2014 Sep 8;5:4675. doi: 10.1038/ncomms5675. Nat Commun. 2014. PMID: 25197941 Free PMC article.
-
Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution?Nutrients. 2015 Aug 17;7(8):6900-23. doi: 10.3390/nu7085314. Nutrients. 2015. PMID: 26287240 Free PMC article. Review.
-
Celiac disease and autoimmunity: review and controversies.Curr Allergy Asthma Rep. 2013 Aug;13(4):347-53. doi: 10.1007/s11882-013-0352-1. Curr Allergy Asthma Rep. 2013. PMID: 23681421 Free PMC article. Review.
-
Improving coeliac disease risk prediction by testing non-HLA variants additional to HLA variants.Gut. 2014 Mar;63(3):415-22. doi: 10.1136/gutjnl-2012-304110. Epub 2013 May 23. Gut. 2014. PMID: 23704318 Free PMC article. Clinical Trial.
References
-
- Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D'alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D'hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppä V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Rückert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sørensen PS, Søndergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvänen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A, Consortium International Multiple Sclerosis Genetics Consortium. Wellcome Trust Case Control Consortium 2 Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214–219. doi: 10.1038/nature10251. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
