

Docking and scoring with ICM: the benchmarking results and strategies for improvement
- PMID: 22569591
- PMCID: PMC3398187
- DOI: 10.1007/s10822-012-9547-0
Docking and scoring with ICM: the benchmarking results and strategies for improvement
Abstract
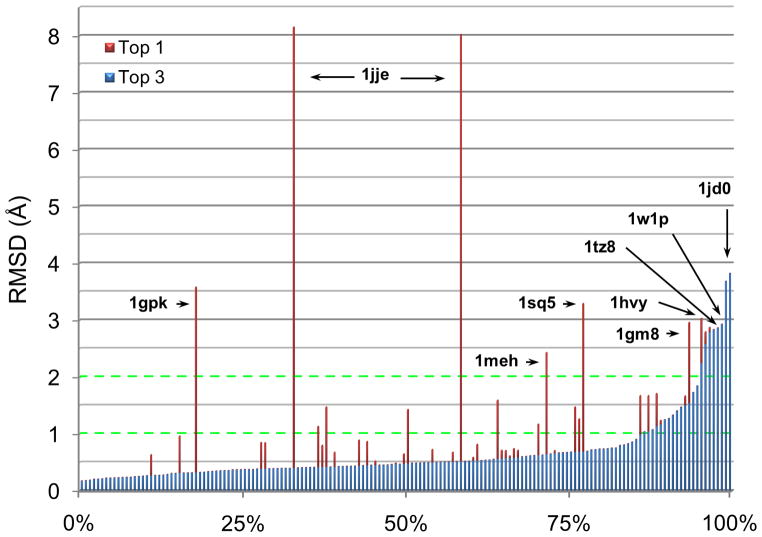
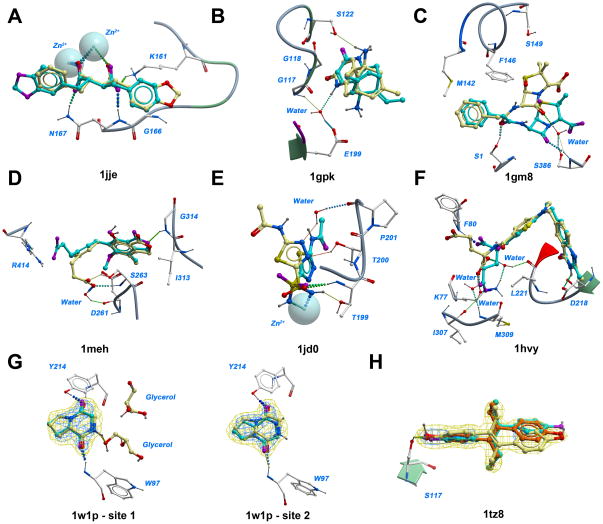
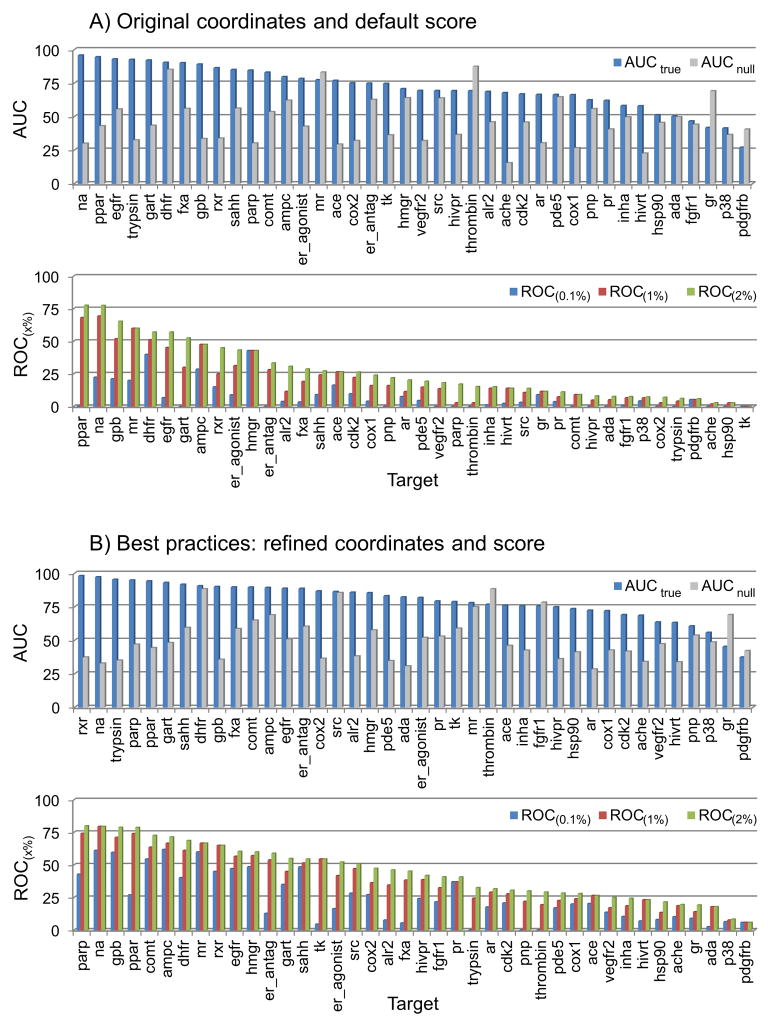
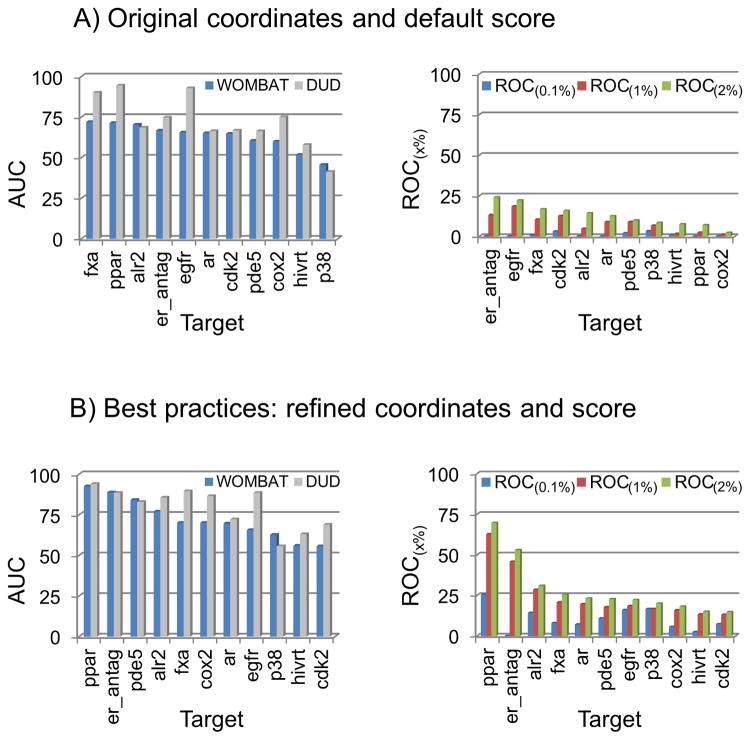
Flexible docking and scoring using the internal coordinate mechanics software (ICM) was benchmarked for ligand binding mode prediction against the 85 co-crystal structures in the modified Astex data set. The ICM virtual ligand screening was tested against the 40 DUD target benchmarks and 11-target WOMBAT sets. The self-docking accuracy was evaluated for the top 1 and top 3 scoring poses at each ligand binding site with near native conformations below 2 Å RMSD found in 91 and 95% of the predictions, respectively. The virtual ligand screening using single rigid pocket conformations provided the median area under the ROC curves equal to 69.4 with 22.0% true positives recovered at 2% false positive rate. Significant improvements up to ROC AUC = 82.2 and ROC((2%)) = 45.2 were achieved following our best practices for flexible pocket refinement and out-of-pocket binding rescore. The virtual screening can be further improved by considering multiple conformations of the target.
Figures
Similar articles
-
Docking performance of the glide program as evaluated on the Astex and DUD datasets: a complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide.J Comput Aided Mol Des. 2012 Jun;26(6):787-99. doi: 10.1007/s10822-012-9575-9. Epub 2012 May 11. J Comput Aided Mol Des. 2012. PMID: 22576241
-
Surflex-Dock: Docking benchmarks and real-world application.J Comput Aided Mol Des. 2012 Jun;26(6):687-99. doi: 10.1007/s10822-011-9533-y. Epub 2012 May 9. J Comput Aided Mol Des. 2012. PMID: 22569590 Free PMC article.
-
Lead Finder docking and virtual screening evaluation with Astex and DUD test sets.J Comput Aided Mol Des. 2012 Jun;26(6):725-35. doi: 10.1007/s10822-012-9549-y. Epub 2012 May 9. J Comput Aided Mol Des. 2012. PMID: 22569592
-
Advancing Ligand Docking through Deep Learning: Challenges and Prospects in Virtual Screening.Acc Chem Res. 2024 May 21;57(10):1500-1509. doi: 10.1021/acs.accounts.4c00093. Epub 2024 Apr 5. Acc Chem Res. 2024. PMID: 38577892 Review.
-
Are predicted protein structures of any value for binding site prediction and virtual ligand screening?Curr Opin Struct Biol. 2013 Apr;23(2):191-7. doi: 10.1016/j.sbi.2013.01.009. Epub 2013 Feb 14. Curr Opin Struct Biol. 2013. PMID: 23415854 Free PMC article. Review.
Cited by
-
Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study.Biomolecules. 2024 Jul 17;14(7):860. doi: 10.3390/biom14070860. Biomolecules. 2024. PMID: 39062574 Free PMC article.
-
Structural Insights into the Mechanisms of Action of Functionally Distinct Classes of Chikungunya Virus Nonstructural Protein 1 Inhibitors.Antimicrob Agents Chemother. 2021 Jun 17;65(7):e0256620. doi: 10.1128/AAC.02566-20. Epub 2021 Jun 17. Antimicrob Agents Chemother. 2021. PMID: 33875421 Free PMC article.
-
In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes.Molecules. 2022 Aug 24;27(17):5400. doi: 10.3390/molecules27175400. Molecules. 2022. PMID: 36080168 Free PMC article.
-
Alchemical Grid Dock (AlGDock): Binding Free Energy Calculations between Flexible Ligands and Rigid Receptors.J Comput Chem. 2020 Mar 15;41(7):715-730. doi: 10.1002/jcc.26036. Epub 2019 Aug 9. J Comput Chem. 2020. PMID: 31397498 Free PMC article.
-
Fast docking on graphics processing units via Ray-Casting.PLoS One. 2013 Aug 16;8(8):e70661. doi: 10.1371/journal.pone.0070661. eCollection 2013. PLoS One. 2013. PMID: 23976948 Free PMC article.
References
-
- Andricopulo AD, Salum LB, Abraham DJ. Structure-based drug design strategies in medicinal chemistry. Curr Top Med Chem. 2009;9:771–790. - PubMed
-
- Kroemer RT. Structure-based drug design: Docking and scoring. Curr Protein Peptide Sci. 2007;8:312–328. - PubMed
-
- Morra G, Genoni A, Neves MAC, Merz KM, Colombo G. Molecular recognition and drug-lead identification: What can molecular simulations tell us? Curr Med Chem. 2010;17:25–41. - PubMed
-
- Zou XQ, Sun YX, Kuntz ID. Inclusion of solvation in ligand binding free energy calculations using the generalized-born model. J Am Chem Soc. 1999;121:8033–8043.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
