

Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans
- PMID: 22560997
- PMCID: PMC3445719
- DOI: 10.1016/j.neuro.2012.04.019
Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans
Abstract
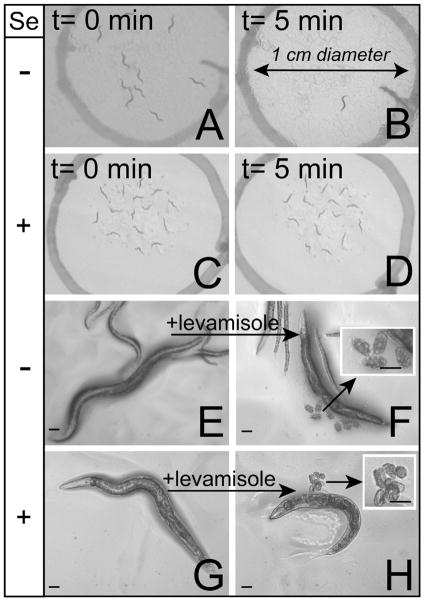
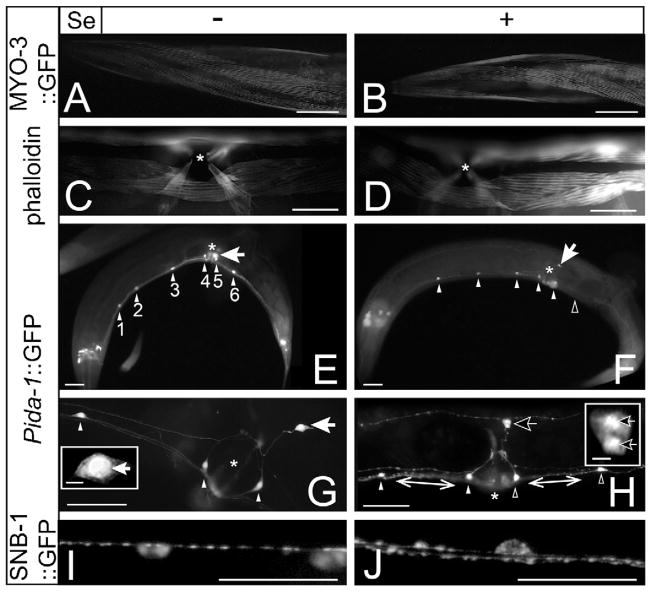
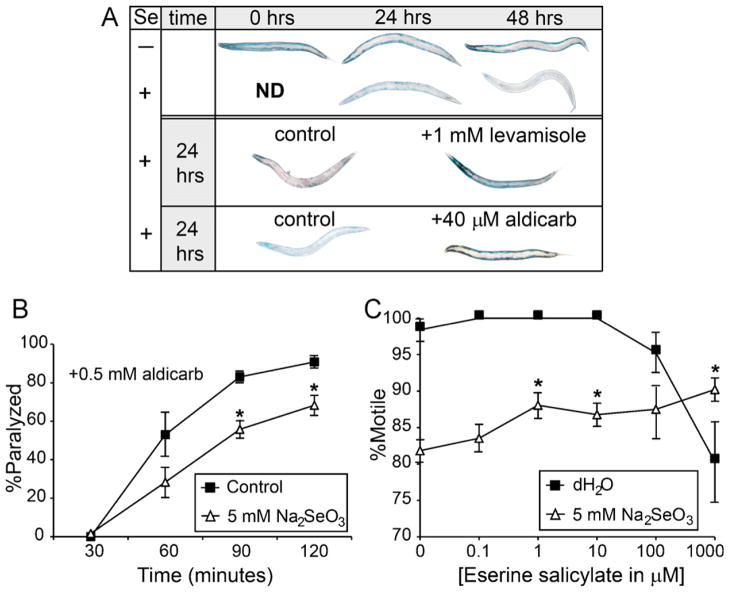
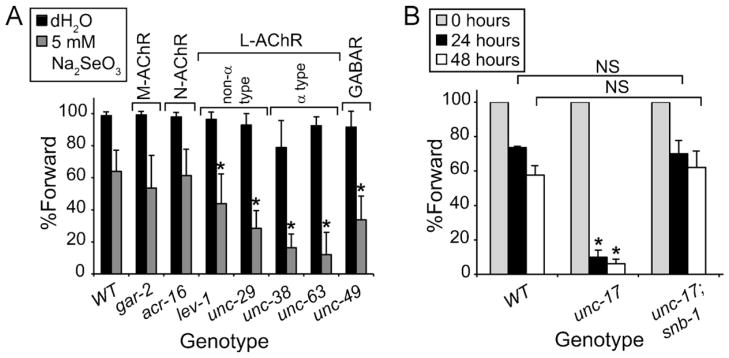
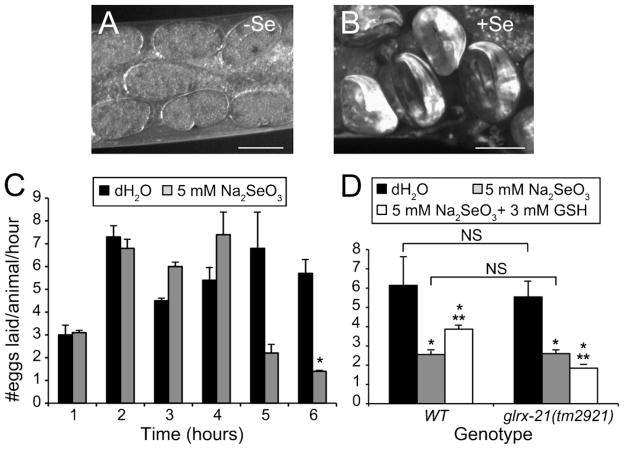
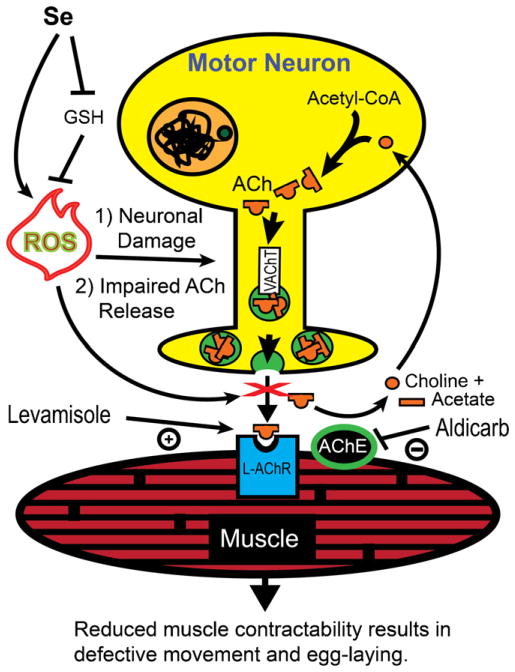
Selenium is an essential micronutrient required for cellular antioxidant systems, yet at higher doses it induces oxidative stress. Additionally, in vertebrates environmental exposures to toxic levels of selenium can cause paralysis and death. Here we show that selenium-induced oxidative stress leads to decreased cholinergic signaling and degeneration of cholinergic neurons required for movement and egg-laying in Caenorhabditis elegans. Exposure to high levels of selenium leads to proteolysis of a soluble muscle protein through mechanisms suppressible by two pharmacological agents, levamisole and aldicarb which enhance cholinergic signaling in muscle. In addition, animals with reduction-of-function mutations in genes encoding post-synaptic levamisole-sensitive acetylcholine receptor subunits or the vesicular acetylcholine transporter developed impaired forward movement faster during selenium-exposure than normal animals, again confirming that selenium reduces cholinergic signaling. Finally, the antioxidant reduced glutathione, inhibits selenium-induced reductions in egg-laying through a cellular protective mechanism dependent on the C. elegans glutaredoxin, GLRX-21. These studies provide evidence that the environmental toxicant selenium induces neurodegeneration of cholinergic neurons through depletion of glutathione, a mechanism linked to the neuropathology of Alzheimer's disease, amyotrophic lateral sclerosis, and Parkinson's disease.
Copyright © 2012 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
Similar articles
-
The neurodegenerative effects of selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans.Neurotoxicology. 2014 Mar;41(100):28-43. doi: 10.1016/j.neuro.2013.12.012. Epub 2014 Jan 6. Neurotoxicology. 2014. PMID: 24406377 Free PMC article.
-
The glutaredoxin GLRX-21 functions to prevent selenium-induced oxidative stress in Caenorhabditis elegans.Toxicol Sci. 2010 Dec;118(2):530-43. doi: 10.1093/toxsci/kfq273. Epub 2010 Sep 10. Toxicol Sci. 2010. PMID: 20833709 Free PMC article.
-
A transmembrane protein required for acetylcholine receptor clustering in Caenorhabditis elegans.Nature. 2004 Sep 30;431(7008):578-82. doi: 10.1038/nature02893. Nature. 2004. PMID: 15457263 Free PMC article.
-
Role of selenium toxicity and oxidative stress in aquatic birds.Aquat Toxicol. 2002 Apr;57(1-2):11-26. doi: 10.1016/s0166-445x(01)00263-6. Aquat Toxicol. 2002. PMID: 11879935 Review.
-
Watching worms whither: modeling neurodegeneration in C. elegans.Prog Mol Biol Transl Sci. 2011;100:499-514. doi: 10.1016/B978-0-12-384878-9.00015-7. Prog Mol Biol Transl Sci. 2011. PMID: 21377635 Free PMC article. Review.
Cited by
-
Deep learning-enabled analysis reveals distinct neuronal phenotypes induced by aging and cold-shock.BMC Biol. 2020 Sep 23;18(1):130. doi: 10.1186/s12915-020-00861-w. BMC Biol. 2020. PMID: 32967665 Free PMC article.
-
Selenium species-dependent toxicity, bioavailability and metabolic transformations in Caenorhabditis elegans.Metallomics. 2018 Jun 20;10(6):818-827. doi: 10.1039/c8mt00066b. Metallomics. 2018. PMID: 29770420 Free PMC article.
-
Stem Cells as Potential Targets of Polyphenols in Multiple Sclerosis and Alzheimer's Disease.Biomed Res Int. 2018 Jul 12;2018:1483791. doi: 10.1155/2018/1483791. eCollection 2018. Biomed Res Int. 2018. PMID: 30112360 Free PMC article. Review.
-
Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases.Antioxid Redox Signal. 2014 Jan 10;20(2):217-35. doi: 10.1089/ars.2012.5051. Epub 2013 Jul 3. Antioxid Redox Signal. 2014. PMID: 23641861 Free PMC article.
-
HIF-1 Has a Central Role in Caenorhabditis elegans Organismal Response to Selenium.Front Genet. 2020 Feb 25;11:63. doi: 10.3389/fgene.2020.00063. eCollection 2020. Front Genet. 2020. PMID: 32161616 Free PMC article.
References
-
- Alfonso A, Grundahl K, Duerr JS, Han HP, Rand JB. The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science. 1993;261(5121):617–9. - PubMed
-
- Bonilla-Ramirez L, Jimenez-Del-Rio M, Velez-Pardo C. Acute and chronic metal exposure impairs locomotion activity in Drosophila melanogaster: a model to study Parkinsonism. Biometals. 2011;24(6):1045–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
