

Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro
- PMID: 22527638
- PMCID: PMC3368277
- DOI: 10.1007/s10974-012-9292-y
Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro
Abstract
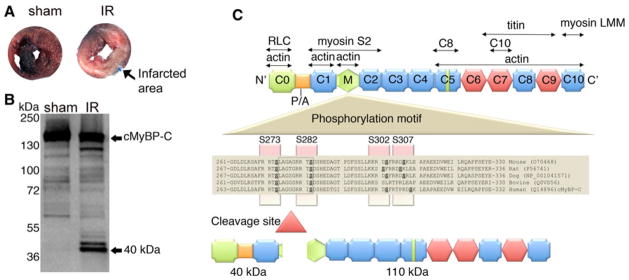
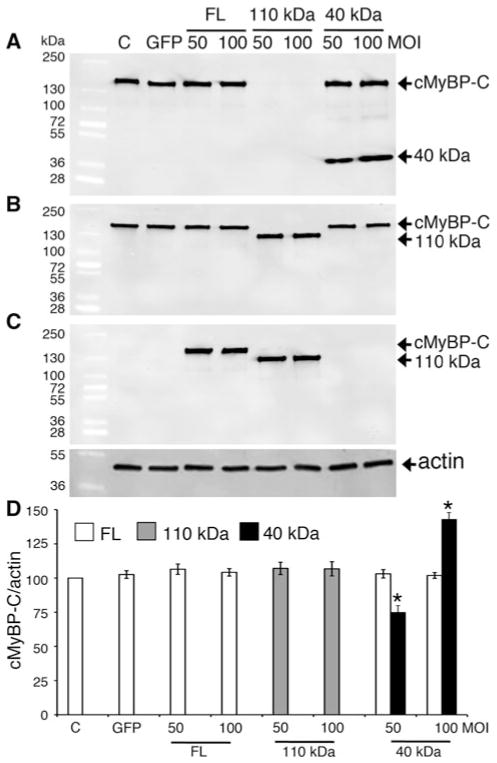
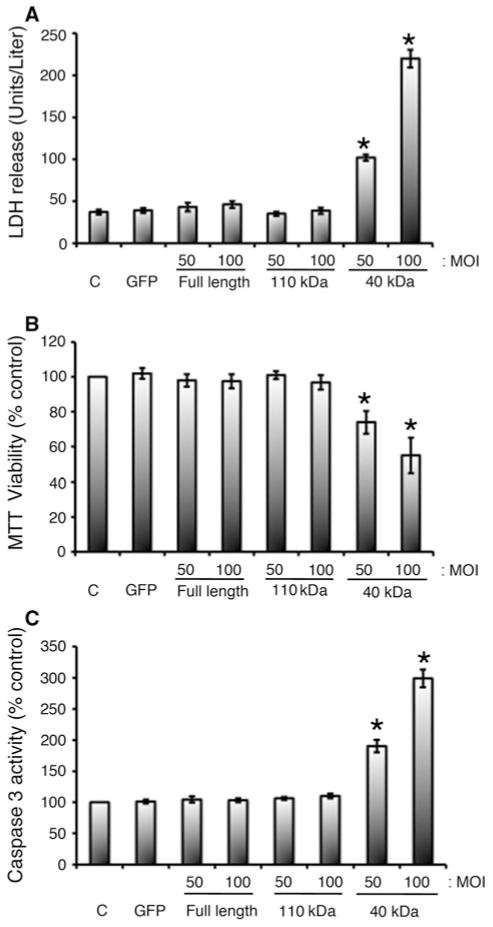
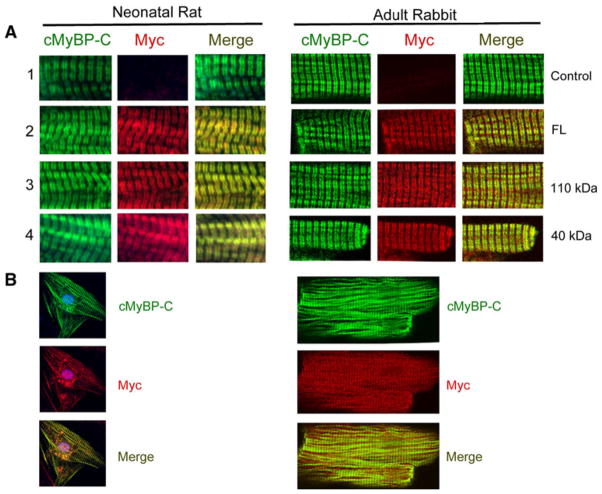
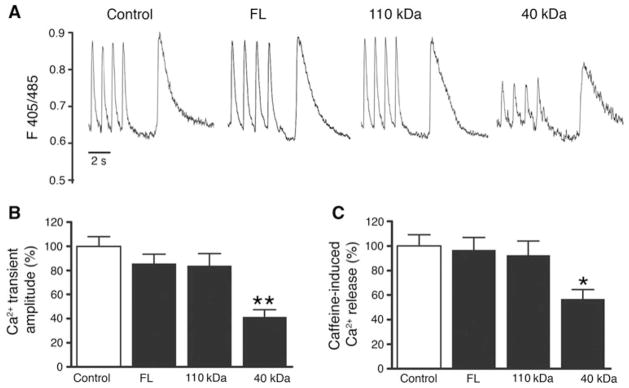
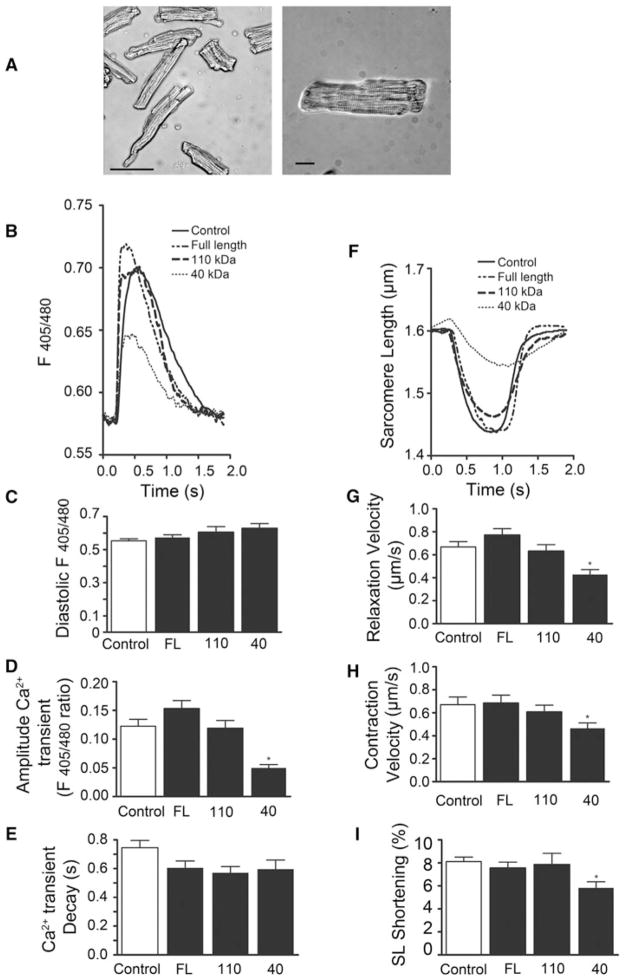
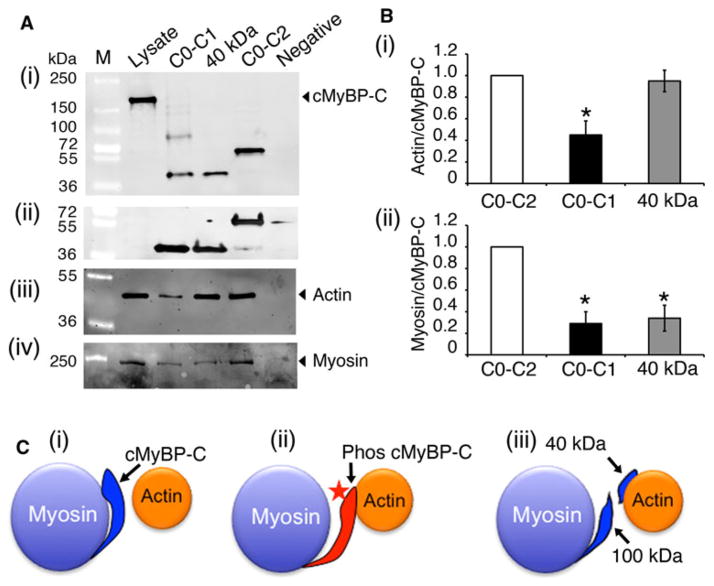
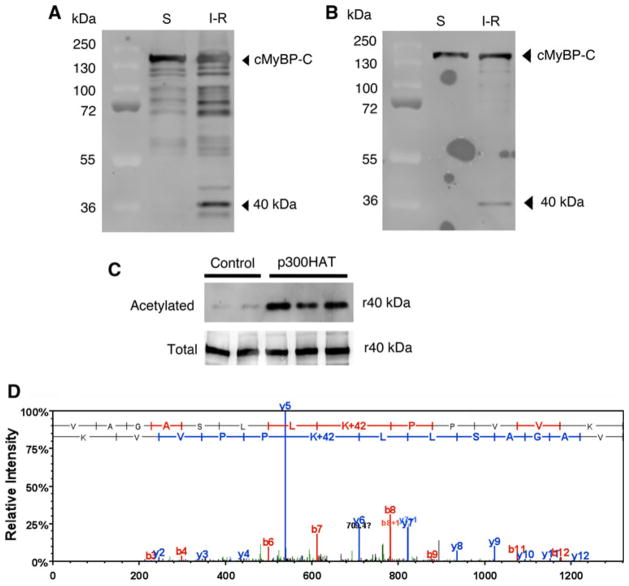
Cardiac myosin binding protein-C (cMyBP-C) plays a role in sarcomeric structure and stability, as well as modulating heart muscle contraction. The 150 kDa full-length (FL) cMyBP-C has been shown to undergo proteolytic cleavage during ischemia-reperfusion injury, producing an N-terminal 40 kDa fragment (mass 29 kDa) that is predominantly associated with post-ischemic contractile dysfunction. Thus far, the pathogenic properties of such truncated cMyBP-C proteins have not been elucidated. In the present study, we hypothesized that the presence of these 40 kDa fragments is toxic to cardiomyocytes, compared to the 110 kDa C-terminal fragment and FL cMyBP-C. To test this hypothesis, we infected neonatal rat ventricular cardiomyocytes and adult rabbit ventricular cardiomyocytes with adenoviruses expressing the FL, 110 and 40 kDa fragments of cMyBP-C, and measured cytotoxicity, Ca(2+) transients, contractility, and protein-protein interactions. Here we show that expression of 40 kDa fragments in neonatal rat ventricular cardiomyocytes significantly increases LDH release and caspase 3 activity, significantly reduces cell viability, and impairs Ca(2+) handling. Adult cardiomyocytes expressing 40 kDa fragments exhibited similar impairment of Ca(2+) handling along with a significant reduction of sarcomere length shortening, relaxation velocity, and contraction velocity. Pull-down assays using recombinant proteins showed that the 40 kDa fragment binds significantly to sarcomeric actin, comparable to C0-C2 domains. In addition, we discovered several acetylation sites within the 40 kDa fragment that could potentially affect actomyosin function. Altogether, our data demonstrate that the 40 kDa cleavage fragments of cMyBP-C are toxic to cardiomyocytes and significantly impair contractility and Ca(2+) handling via inhibition of actomyosin function. By elucidating the deleterious effects of endogenously expressed cMyBP-C N-terminal fragments on sarcomere function, these data contribute to the understanding of contractile dysfunction following myocardial injury.
Figures
Similar articles
-
Amino terminus of cardiac myosin binding protein-C regulates cardiac contractility.J Mol Cell Cardiol. 2021 Jul;156:33-44. doi: 10.1016/j.yjmcc.2021.03.009. Epub 2021 Mar 26. J Mol Cell Cardiol. 2021. PMID: 33781820 Free PMC article.
-
Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium.J Biol Chem. 2014 Mar 28;289(13):8818-27. doi: 10.1074/jbc.M113.541128. Epub 2014 Feb 7. J Biol Chem. 2014. PMID: 24509847 Free PMC article.
-
COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes.J Mol Biol. 1999 Nov 26;294(2):443-56. doi: 10.1006/jmbi.1999.3276. J Mol Biol. 1999. PMID: 10610770
-
Phosphorylation and function of cardiac myosin binding protein-C in health and disease.J Mol Cell Cardiol. 2010 May;48(5):866-75. doi: 10.1016/j.yjmcc.2009.11.014. Epub 2009 Dec 3. J Mol Cell Cardiol. 2010. PMID: 19962384 Free PMC article. Review.
-
Species-specific differences in the Pro-Ala rich region of cardiac myosin binding protein-C.J Muscle Res Cell Motil. 2009 Dec;30(7-8):303-6. doi: 10.1007/s10974-010-9207-8. Epub 2010 Mar 9. J Muscle Res Cell Motil. 2009. PMID: 20217194 Free PMC article. Review.
Cited by
-
Amino terminus of cardiac myosin binding protein-C regulates cardiac contractility.J Mol Cell Cardiol. 2021 Jul;156:33-44. doi: 10.1016/j.yjmcc.2021.03.009. Epub 2021 Mar 26. J Mol Cell Cardiol. 2021. PMID: 33781820 Free PMC article.
-
An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure.Circ Res. 2013 Aug 16;113(5):553-61. doi: 10.1161/CIRCRESAHA.113.301225. Epub 2013 Jul 12. Circ Res. 2013. PMID: 23852539 Free PMC article.
-
Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology.Gene. 2015 Dec 1;573(2):188-97. doi: 10.1016/j.gene.2015.09.008. Epub 2015 Sep 8. Gene. 2015. PMID: 26358504 Free PMC article. Review.
-
Increase in cardiac myosin binding protein-C plasma levels is a sensitive and cardiac-specific biomarker of myocardial infarction.Am J Cardiovasc Dis. 2013 Jun 10;3(2):60-70. Print 2013. Am J Cardiovasc Dis. 2013. PMID: 23785583 Free PMC article.
-
Etiology of genetic muscle disorders induced by mutations in fast and slow skeletal MyBP-C paralogs.Exp Mol Med. 2023 Mar;55(3):502-509. doi: 10.1038/s12276-023-00953-x. Epub 2023 Mar 1. Exp Mol Med. 2023. PMID: 36854776 Free PMC article. Review.
References
-
- Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J, Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J Mol Cell Cardiol. 2010;49:1003–1011. - PubMed
-
- Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906–912. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
