

PSORS2 is due to mutations in CARD14
- PMID: 22521418
- PMCID: PMC3376640
- DOI: 10.1016/j.ajhg.2012.03.012
PSORS2 is due to mutations in CARD14
Abstract
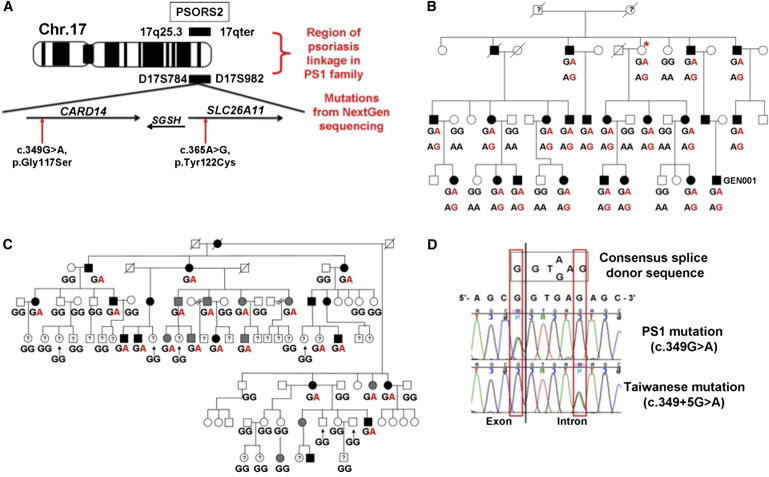
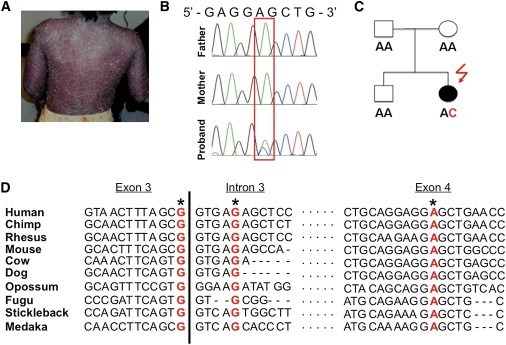
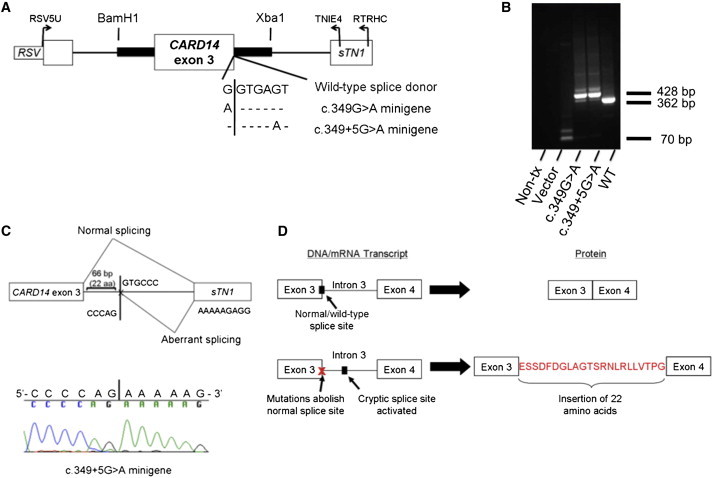
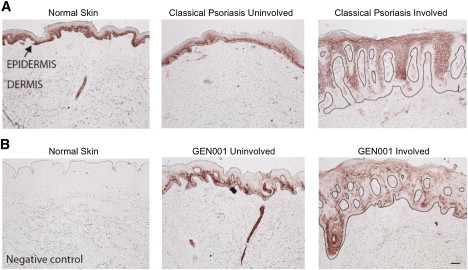
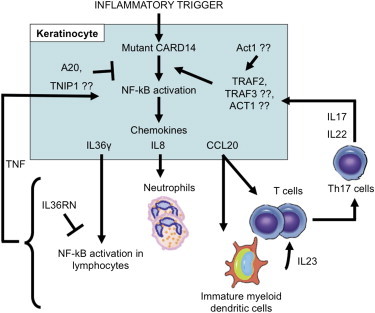
Psoriasis is a common, immune-mediated genetic disorder of the skin and is associated with arthritis in approximately 30% of cases. Previously, we localized PSORS2 (psoriasis susceptibility locus 2) to chromosomal region 17q25.3-qter after a genome-wide linkage scan in a family of European ancestry with multiple cases of psoriasis and psoriatic arthritis. Linkage to PSORS2 was also observed in a Taiwanese family with multiple psoriasis-affected members. In caspase recruitment domain family, member 14 (CARD14), we identified unique gain-of-function mutations that segregated with psoriasis by using genomic capture and DNA sequencing. The mutations c.349G>A (p.Gly117Ser) (in the family of European descent) and c.349+5G>A (in the Taiwanese family) altered splicing between CARD14 exons 3 and 4. A de novo CARD14 mutation, c.413A>C (p.Glu138Ala), was detected in a child with sporadic, early-onset, generalized pustular psoriasis. CARD14 activates nuclear factor kappa B (NF-kB), and compared with wild-type CARD14, the p.Gly117Ser and p.Glu138Ala substitutions were shown to lead to enhanced NF-kB activation and upregulation of a subset of psoriasis-associated genes in keratinocytes. These genes included chemokine (C-C motif) ligand 20 (CCL20) and interleukin 8 (IL8). CARD14 is localized mainly in the basal and suprabasal layers of healthy skin epidermis, whereas in lesional psoriatic skin, it is reduced in the basal layer and more diffusely upregulated in the suprabasal layers of the epidermis. We propose that, after a triggering event that can include epidermal injury, rare gain-of-function mutations in CARD14 initiate a process that includes inflammatory cell recruitment by keratinocytes. This perpetuates a vicious cycle of epidermal inflammation and regeneration, a cycle which is the hallmark of psoriasis.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis.Am J Hum Genet. 2012 May 4;90(5):796-808. doi: 10.1016/j.ajhg.2012.03.013. Epub 2012 Apr 19. Am J Hum Genet. 2012. PMID: 22521419 Free PMC article.
-
CARD14 alterations in Tunisian patients with psoriasis and further characterization in European cohorts.Br J Dermatol. 2016 Feb;174(2):330-7. doi: 10.1111/bjd.14158. Epub 2015 Nov 17. Br J Dermatol. 2016. PMID: 26358359 Free PMC article.
-
CARD14 expression in dermal endothelial cells in psoriasis.PLoS One. 2014 Nov 4;9(11):e111255. doi: 10.1371/journal.pone.0111255. eCollection 2014. PLoS One. 2014. PMID: 25369198 Free PMC article.
-
Clinical and Genetic Heterogeneity of CARD14 Mutations in Psoriatic Skin Disease.Front Immunol. 2018 Oct 16;9:2239. doi: 10.3389/fimmu.2018.02239. eCollection 2018. Front Immunol. 2018. PMID: 30386326 Free PMC article. Review.
-
CARD14-Mediated Activation of Paracaspase MALT1 in Keratinocytes: Implications for Psoriasis.J Invest Dermatol. 2017 Mar;137(3):569-575. doi: 10.1016/j.jid.2016.09.031. Epub 2016 Dec 8. J Invest Dermatol. 2017. PMID: 27939769 Review.
Cited by
-
The Chemokine, CCL20, and Its Receptor, CCR6, in the Pathogenesis and Treatment of Psoriasis and Psoriatic Arthritis.J Psoriasis Psoriatic Arthritis. 2023 Jul;8(3):107-117. doi: 10.1177/24755303231159106. Epub 2023 Mar 12. J Psoriasis Psoriatic Arthritis. 2023. PMID: 39296310 Free PMC article. Review.
-
A Case of Old Age-Onset Generalized Pustular Psoriasis with a Deficiency of IL-36RN (DITRA) Treated by Granulocyte and Monocyte Apheresis.Case Rep Dermatol. 2015 Feb 21;7(1):29-35. doi: 10.1159/000380876. eCollection 2015 Jan-Apr. Case Rep Dermatol. 2015. PMID: 25848350 Free PMC article.
-
The immunogenetics of Psoriasis: A comprehensive review.J Autoimmun. 2015 Nov;64:66-73. doi: 10.1016/j.jaut.2015.07.008. Epub 2015 Jul 26. J Autoimmun. 2015. PMID: 26215033 Free PMC article. Review.
-
Analysis of Gastric Cancer Transcriptome Allows the Identification of Histotype Specific Molecular Signatures With Prognostic Potential.Front Oncol. 2021 May 3;11:663771. doi: 10.3389/fonc.2021.663771. eCollection 2021. Front Oncol. 2021. PMID: 34012923 Free PMC article.
-
From bench to bedside and back again: translational research in autoinflammation.Nat Rev Rheumatol. 2015 Oct;11(10):573-85. doi: 10.1038/nrrheum.2015.79. Epub 2015 Jun 16. Nat Rev Rheumatol. 2015. PMID: 26077920 Review.
References
-
- Lowes M.A., Bowcock A.M., Krueger J.G. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AR050266/AR/NIAMS NIH HHS/United States
- T32HL083822/HL/NHLBI NIH HHS/United States
- T32 AR007279/AR/NIAMS NIH HHS/United States
- T32AR007279/AR/NIAMS NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
- T32HG000045/HG/NHGRI NIH HHS/United States
- AR050266/AR/NIAMS NIH HHS/United States
- T32 HL083822/HL/NHLBI NIH HHS/United States
- 5RC1AR058681/AR/NIAMS NIH HHS/United States
- T32 GM007200/GM/NIGMS NIH HHS/United States
- AR060222/AR/NIAMS NIH HHS/United States
- T32 HG000045/HG/NHGRI NIH HHS/United States
- T32GM07200/GM/NIGMS NIH HHS/United States
- R01 AR060222/AR/NIAMS NIH HHS/United States
- RC1 AR058681/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
