

Type 2 diabetes risk alleles demonstrate extreme directional differentiation among human populations, compared to other diseases
- PMID: 22511877
- PMCID: PMC3325177
- DOI: 10.1371/journal.pgen.1002621
Type 2 diabetes risk alleles demonstrate extreme directional differentiation among human populations, compared to other diseases
Abstract
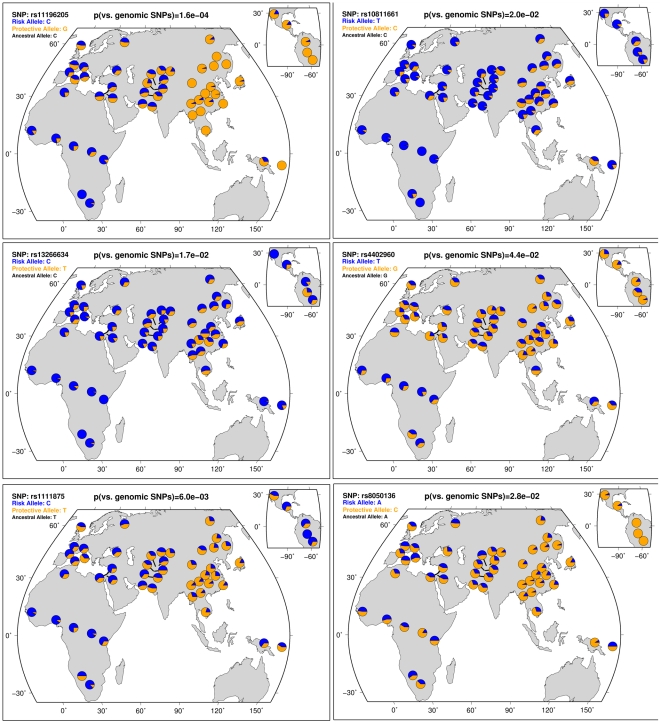
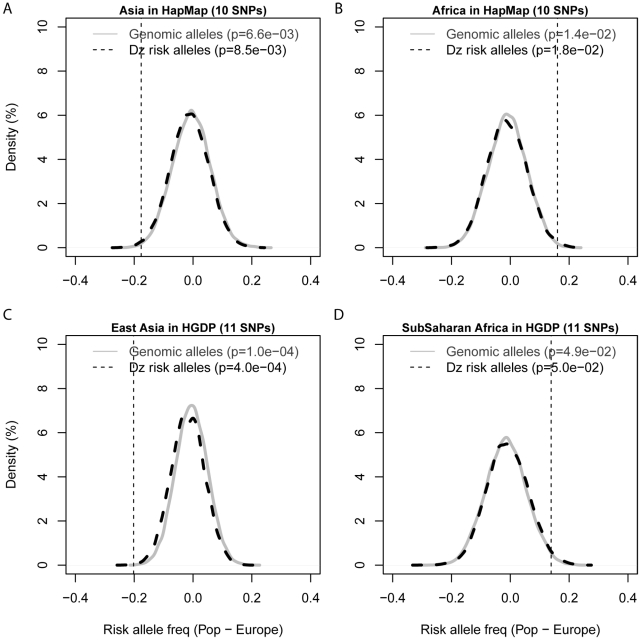
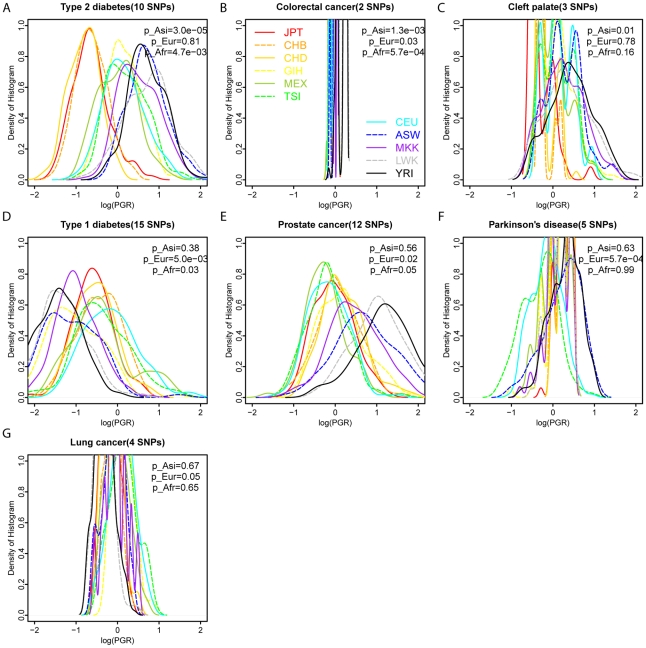
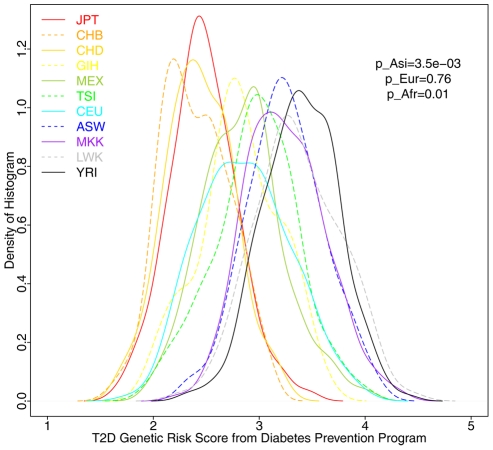
Many disease-susceptible SNPs exhibit significant disparity in ancestral and derived allele frequencies across worldwide populations. While previous studies have examined population differentiation of alleles at specific SNPs, global ethnic patterns of ensembles of disease risk alleles across human diseases are unexamined. To examine these patterns, we manually curated ethnic disease association data from 5,065 papers on human genetic studies representing 1,495 diseases, recording the precise risk alleles and their measured population frequencies and estimated effect sizes. We systematically compared the population frequencies of cross-ethnic risk alleles for each disease across 1,397 individuals from 11 HapMap populations, 1,064 individuals from 53 HGDP populations, and 49 individuals with whole-genome sequences from 10 populations. Type 2 diabetes (T2D) demonstrated extreme directional differentiation of risk allele frequencies across human populations, compared with null distributions of European-frequency matched control genomic alleles and risk alleles for other diseases. Most T2D risk alleles share a consistent pattern of decreasing frequencies along human migration into East Asia. Furthermore, we show that these patterns contribute to disparities in predicted genetic risk across 1,397 HapMap individuals, T2D genetic risk being consistently higher for individuals in the African populations and lower in the Asian populations, irrespective of the ethnicity considered in the initial discovery of risk alleles. We observed a similar pattern in the distribution of T2D Genetic Risk Scores, which are associated with an increased risk of developing diabetes in the Diabetes Prevention Program cohort, for the same individuals. This disparity may be attributable to the promotion of energy storage and usage appropriate to environments and inconsistent energy intake. Our results indicate that the differential frequencies of T2D risk alleles may contribute to the observed disparity in T2D incidence rates across ethnic populations.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





Similar articles
-
Worldwide distribution of the MYH9 kidney disease susceptibility alleles and haplotypes: evidence of historical selection in Africa.PLoS One. 2010 Jul 9;5(7):e11474. doi: 10.1371/journal.pone.0011474. PLoS One. 2010. PMID: 20634883 Free PMC article.
-
Common variants in the TCF7L2 gene and predisposition to type 2 diabetes in UK European Whites, Indian Asians and Afro-Caribbean men and women.J Mol Med (Berl). 2006 Dec;84(12):1005-14. doi: 10.1007/s00109-006-0108-7. J Mol Med (Berl). 2006. PMID: 17665514
-
A comparison of type 2 diabetes risk allele load between African Americans and European Americans.Hum Genet. 2014 Dec;133(12):1487-95. doi: 10.1007/s00439-014-1486-5. Epub 2014 Oct 2. Hum Genet. 2014. PMID: 25273842 Free PMC article.
-
Progress in Defining the Genetic Contribution to Type 2 Diabetes in Individuals of East Asian Ancestry.Curr Diab Rep. 2021 Apr 13;21(6):17. doi: 10.1007/s11892-021-01388-2. Curr Diab Rep. 2021. PMID: 33846905 Review.
-
Genetic susceptibility factors of Type 1 diabetes in Asians.Diabetes Metab Res Rev. 2001 Jan-Feb;17(1):2-11. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr164>3.0.co;2-m. Diabetes Metab Res Rev. 2001. PMID: 11241886 Review.
Cited by
-
Marine N-3 polyunsaturated fatty acids are inversely associated with risk of type 2 diabetes in Asians: a systematic review and meta-analysis.PLoS One. 2012;7(9):e44525. doi: 10.1371/journal.pone.0044525. Epub 2012 Sep 11. PLoS One. 2012. PMID: 22984522 Free PMC article. Review.
-
Type 2 diabetes mellitus: distribution of genetic markers in Kazakh population.Clin Interv Aging. 2018 Mar 5;13:377-388. doi: 10.2147/CIA.S156044. eCollection 2018. Clin Interv Aging. 2018. PMID: 29551892 Free PMC article.
-
The potential of novel biomarkers to improve risk prediction of type 2 diabetes.Diabetologia. 2014 Jan;57(1):16-29. doi: 10.1007/s00125-013-3061-3. Diabetologia. 2014. PMID: 24078135 Review.
-
Revisiting the thrifty gene hypothesis via 65 loci associated with susceptibility to type 2 diabetes.Am J Hum Genet. 2014 Feb 6;94(2):176-85. doi: 10.1016/j.ajhg.2013.12.010. Epub 2014 Jan 9. Am J Hum Genet. 2014. PMID: 24412096 Free PMC article.
-
The melatonin receptor 1B gene links circadian rhythms and type 2 diabetes mellitus: an evolutionary story.Ann Med. 2023 Dec;55(1):1262-1286. doi: 10.1080/07853890.2023.2191218. Ann Med. 2023. PMID: 36974476 Free PMC article. Review.
References
-
- van Dieren S, Beulens JW, van der Schouw YT, Grobbee DE, Neal B. The global burden of diabetes and its complications: an emerging pandemic. Eur J Cardiovasc Prev Rehabil. 2010;17(Suppl 1):S3–8. - PubMed
-
- Groop L, Lyssenko V. Genes and type 2 diabetes mellitus. Curr Diab Rep. 2008;8:192–197. - PubMed
-
- Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. - PubMed
-
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
