

The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity
- PMID: 22460905
- PMCID: PMC3320027
- DOI: 10.1038/nature11003
The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity
Erratum in
- Nature. 2012 Dec 13;492(7428):290
-
Addendum: The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity.Nature. 2019 Jan;565(7738):E5-E6. doi: 10.1038/s41586-018-0722-x. Nature. 2019. PMID: 30559381 No abstract available.
Abstract
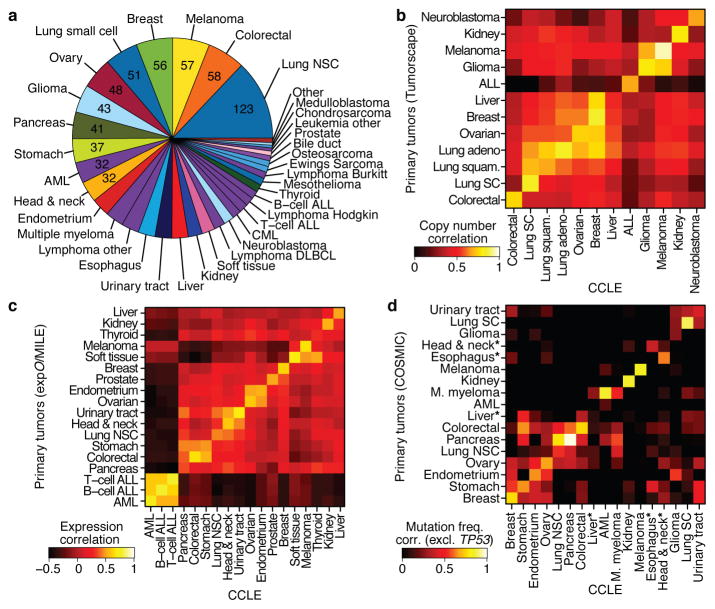
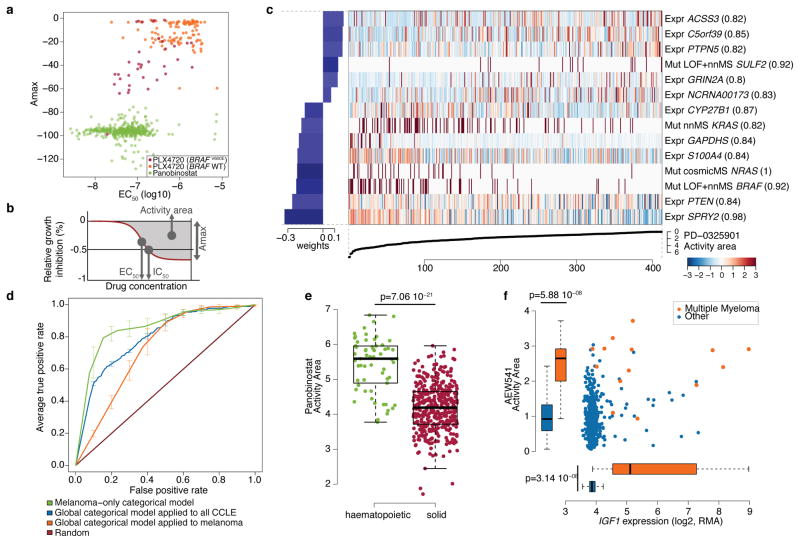
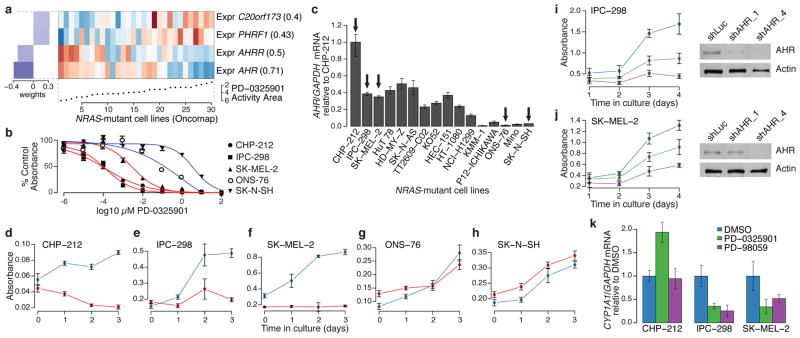
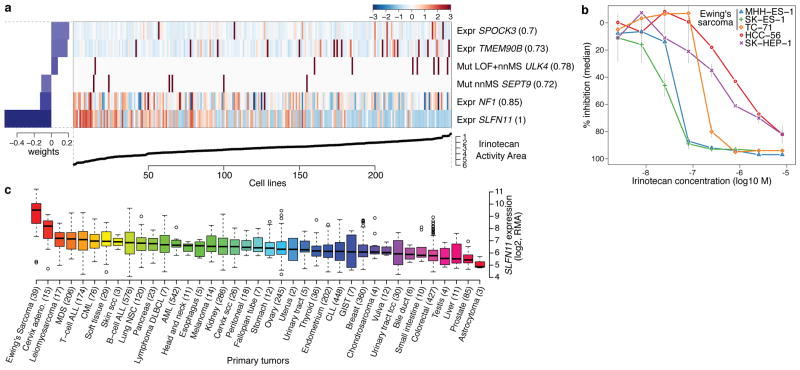
The systematic translation of cancer genomic data into knowledge of tumour biology and therapeutic possibilities remains challenging. Such efforts should be greatly aided by robust preclinical model systems that reflect the genomic diversity of human cancers and for which detailed genetic and pharmacological annotation is available. Here we describe the Cancer Cell Line Encyclopedia (CCLE): a compilation of gene expression, chromosomal copy number and massively parallel sequencing data from 947 human cancer cell lines. When coupled with pharmacological profiles for 24 anticancer drugs across 479 of the cell lines, this collection allowed identification of genetic, lineage, and gene-expression-based predictors of drug sensitivity. In addition to known predictors, we found that plasma cell lineage correlated with sensitivity to IGF1 receptor inhibitors; AHR expression was associated with MEK inhibitor efficacy in NRAS-mutant lines; and SLFN11 expression predicted sensitivity to topoisomerase inhibitors. Together, our results indicate that large, annotated cell-line collections may help to enable preclinical stratification schemata for anticancer agents. The generation of genetic predictions of drug response in the preclinical setting and their incorporation into cancer clinical trial design could speed the emergence of 'personalized' therapeutic regimens.
Conflict of interest statement
Multiple authors are employees of Novartis, Inc., as noted in the affiliations. T.R.G., M.M., and L.A.G. are consultants for and equity holders in Foundation Medicine, Inc. M.M. and L.A.G. are consultants for and receive sponsored research from Novartis, Inc.
Figures
Comment in
-
Drug discovery: Cell lines battle cancer.Nature. 2012 Mar 28;483(7391):544-5. doi: 10.1038/483544a. Nature. 2012. PMID: 22460893 No abstract available.
-
Genetics: Cells line up to be characterized.Nat Rev Clin Oncol. 2012 Apr 3;9(5):249. doi: 10.1038/nrclinonc.2012.56. Nat Rev Clin Oncol. 2012. PMID: 22473099 No abstract available.
-
Cancer genomics: Constructing a 'cancerpaedia'.Nat Rev Genet. 2012 Apr 18;13(5):300. doi: 10.1038/nrg3221. Nat Rev Genet. 2012. PMID: 22510763 No abstract available.
-
Genomics: Constructing a 'cancerpaedia'.Nat Rev Cancer. 2012 Apr 19;12(5):315. doi: 10.1038/nrc3275. Nat Rev Cancer. 2012. PMID: 22513402 No abstract available.
-
Cancer genomics: Constructing a 'cancerpaedia'.Nat Rev Drug Discov. 2012 Apr 30;11(5):353. doi: 10.1038/nrd3730. Nat Rev Drug Discov. 2012. PMID: 22543463 No abstract available.
Similar articles
-
Drug discovery: Cell lines battle cancer.Nature. 2012 Mar 28;483(7391):544-5. doi: 10.1038/483544a. Nature. 2012. PMID: 22460893 No abstract available.
-
Predicting breast cancer drug response using a multiple-layer cell line drug response network model.BMC Cancer. 2021 May 31;21(1):648. doi: 10.1186/s12885-021-08359-6. BMC Cancer. 2021. PMID: 34059012 Free PMC article.
-
Drug-Sensitivity Screening and Genomic Characterization of 45 HPV-Negative Head and Neck Carcinoma Cell Lines for Novel Biomarkers of Drug Efficacy.Mol Cancer Ther. 2018 Sep;17(9):2060-2071. doi: 10.1158/1535-7163.MCT-17-0733. Epub 2018 Jul 3. Mol Cancer Ther. 2018. PMID: 29970484
-
Rapid identification and validation of novel targeted approaches for Glioblastoma: A combined ex vivo-in vivo pharmaco-omic model.Exp Neurol. 2018 Jan;299(Pt B):281-288. doi: 10.1016/j.expneurol.2017.09.006. Epub 2017 Sep 18. Exp Neurol. 2018. PMID: 28923369 Review.
-
Using drug response data to identify molecular effectors, and molecular "omic" data to identify candidate drugs in cancer.Hum Genet. 2015 Jan;134(1):3-11. doi: 10.1007/s00439-014-1482-9. Epub 2014 Sep 12. Hum Genet. 2015. PMID: 25213708 Free PMC article. Review.
Cited by
-
Harnessing the power of AI in precision medicine: NGS-based therapeutic insights for colorectal cancer cohort.Front Oncol. 2024 Oct 7;14:1407465. doi: 10.3389/fonc.2024.1407465. eCollection 2024. Front Oncol. 2024. PMID: 39435285 Free PMC article.
-
Identification of novel prognostic biomarkers by integrating multi-omics data in gastric cancer.BMC Cancer. 2021 Apr 26;21(1):460. doi: 10.1186/s12885-021-08210-y. BMC Cancer. 2021. PMID: 33902514 Free PMC article.
-
Selective targeting of KRAS-driven lung tumorigenesis via unresolved ER stress.JCI Insight. 2021 Apr 8;6(7):e137876. doi: 10.1172/jci.insight.137876. JCI Insight. 2021. PMID: 33830081 Free PMC article.
-
Empowering biologists with multi-omics data: colorectal cancer as a paradigm.Bioinformatics. 2015 May 1;31(9):1436-43. doi: 10.1093/bioinformatics/btu834. Epub 2014 Dec 18. Bioinformatics. 2015. PMID: 25527095 Free PMC article.
-
Cancer cell histone density links global histone acetylation, mitochondrial proteome and histone acetylase inhibitor sensitivity.Commun Biol. 2022 Aug 27;5(1):882. doi: 10.1038/s42003-022-03846-3. Commun Biol. 2022. PMID: 36030322 Free PMC article.
References
-
- Caponigro G, Sellers WR. Advances in the preclinical testing of cancer therapeutic hypotheses. Nat Rev Drug Discov. 2011;10:179–187. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
