

Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis
- PMID: 22454397
- PMCID: PMC3373238
- DOI: 10.1093/hmg/dds116
Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis
Abstract
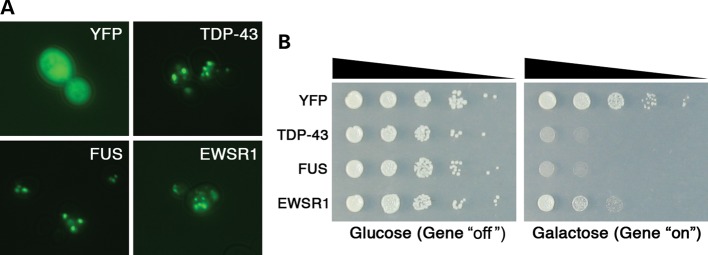
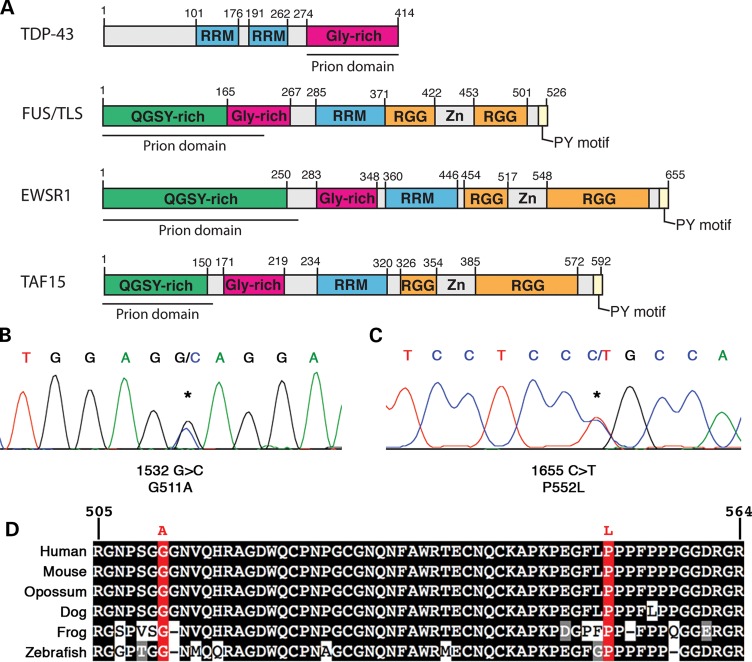
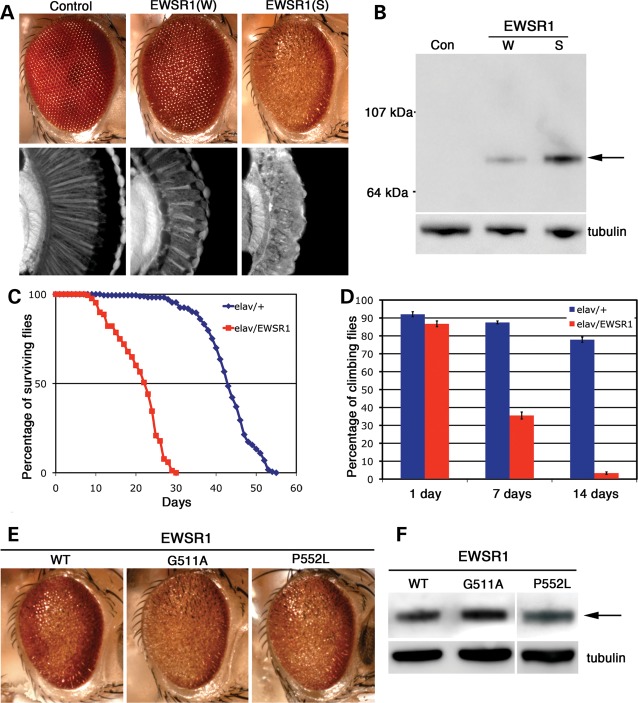
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting motor neurons. Mutations in related RNA-binding proteins TDP-43, FUS/TLS and TAF15 have been connected to ALS. These three proteins share several features, including the presence of a bioinformatics-predicted prion domain, aggregation-prone nature in vitro and in vivo and toxic effects when expressed in multiple model systems. Given these commonalities, we hypothesized that a related protein, EWSR1 (Ewing sarcoma breakpoint region 1), might also exhibit similar properties and therefore could contribute to disease. Here, we report an analysis of EWSR1 in multiple functional assays, including mutational screening in ALS patients and controls. We identified three missense variants in EWSR1 in ALS patients, which were absent in a large number of healthy control individuals. We show that disease-specific variants affect EWSR1 localization in motor neurons. We also provide multiple independent lines of in vitro and in vivo evidence that EWSR1 has similar properties as TDP-43, FUS and TAF15, including aggregation-prone behavior in vitro and ability to confer neurodegeneration in Drosophila. Postmortem analysis of sporadic ALS cases also revealed cytoplasmic mislocalization of EWSR1. Together, our studies highlight a potential role for EWSR1 in ALS, provide a collection of functional assays to be used to assess roles of additional RNA-binding proteins in disease and support an emerging concept that a class of aggregation-prone RNA-binding proteins might contribute broadly to ALS and related neurodegenerative diseases.
Figures



Similar articles
-
RNA-binding proteins with prion-like domains in health and disease.Biochem J. 2017 Apr 7;474(8):1417-1438. doi: 10.1042/BCJ20160499. Biochem J. 2017. PMID: 28389532 Free PMC article. Review.
-
A yeast functional screen predicts new candidate ALS disease genes.Proc Natl Acad Sci U S A. 2011 Dec 27;108(52):20881-90. doi: 10.1073/pnas.1109434108. Epub 2011 Nov 7. Proc Natl Acad Sci U S A. 2011. PMID: 22065782 Free PMC article.
-
Aggregation of FET Proteins as a Pathological Change in Amyotrophic Lateral Sclerosis.Adv Exp Med Biol. 2017;925:1-12. doi: 10.1007/5584_2016_32. Adv Exp Med Biol. 2017. PMID: 27311318 Review.
-
Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses.Nat Commun. 2016 Jul 5;7:12143. doi: 10.1038/ncomms12143. Nat Commun. 2016. PMID: 27378374 Free PMC article.
-
The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease.Brain Res. 2012 Jun 26;1462:61-80. doi: 10.1016/j.brainres.2012.01.016. Epub 2012 Jan 21. Brain Res. 2012. PMID: 22445064 Free PMC article. Review.
Cited by
-
Role and therapeutic potential of liquid-liquid phase separation in amyotrophic lateral sclerosis.J Mol Cell Biol. 2021 Apr 10;13(1):15-28. doi: 10.1093/jmcb/mjaa049. J Mol Cell Biol. 2021. PMID: 32976566 Free PMC article. Review.
-
The Interplay of RNA Binding Proteins, Oxidative Stress and Mitochondrial Dysfunction in ALS.Antioxidants (Basel). 2021 Apr 2;10(4):552. doi: 10.3390/antiox10040552. Antioxidants (Basel). 2021. PMID: 33918215 Free PMC article. Review.
-
Advances in understanding the molecular basis of frontotemporal dementia.Nat Rev Neurol. 2012 Aug;8(8):423-34. doi: 10.1038/nrneurol.2012.117. Epub 2012 Jun 26. Nat Rev Neurol. 2012. PMID: 22732773 Free PMC article. Review.
-
Prion-like proteins: from computational approaches to proteome-wide analysis.FEBS Open Bio. 2021 Sep;11(9):2400-2417. doi: 10.1002/2211-5463.13213. Epub 2021 Jun 17. FEBS Open Bio. 2021. PMID: 34057308 Free PMC article. Review.
-
The function of RNA-binding proteins at the synapse: implications for neurodegeneration.Cell Mol Life Sci. 2015 Oct;72(19):3621-35. doi: 10.1007/s00018-015-1943-x. Epub 2015 Jun 6. Cell Mol Life Sci. 2015. PMID: 26047658 Free PMC article. Review.
References
-
- Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001;2:806–819. doi:10.1038/35097565. - DOI - PubMed
-
- Andersen P.M., Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 2011;7:603–615. doi:10.1038/nrneurol.2011.150. - DOI - PubMed
-
- DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi:10.1016/j.neuron.2011.09.011. - DOI - PMC - PubMed
-
- Deng H.X., Chen W., Hong S.T., Boycott K.M., Gorrie G.H., Siddique N., Yang Y., Fecto F., Shi Y., Zhai H., et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi:10.1038/nature10353. - DOI - PMC - PubMed
-
- Fecto F., Siddique T. Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J. Mol. Neurosci. 2011;45:663–675. doi:10.1007/s12031-011-9637-9. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
