

Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection
- PMID: 22412369
- PMCID: PMC3297584
- DOI: 10.1371/journal.ppat.1002529
Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection
Abstract
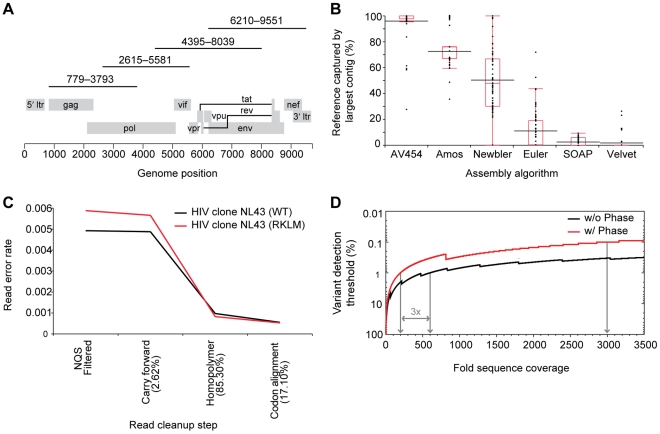
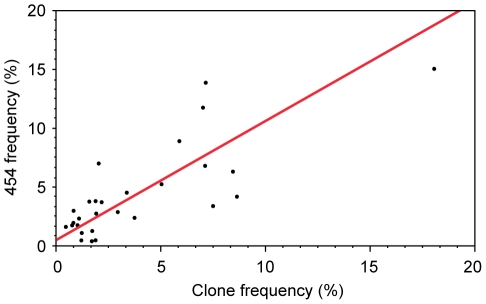
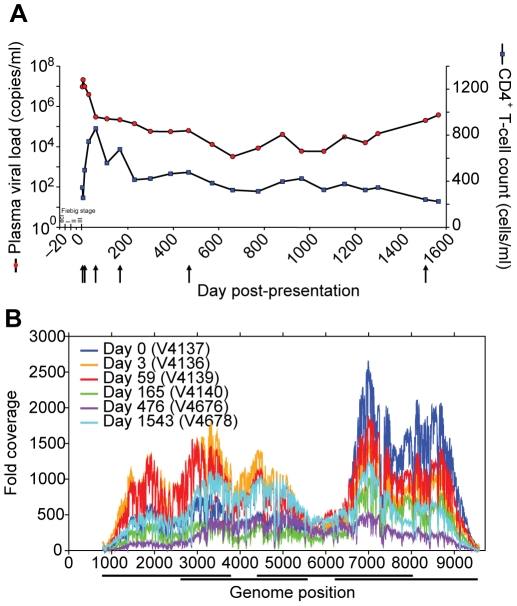
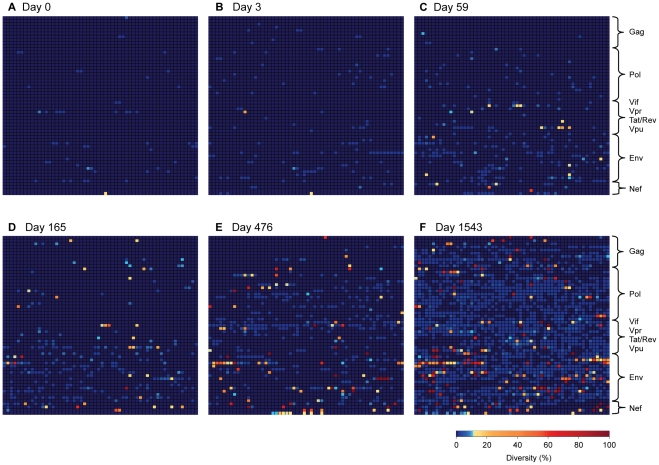
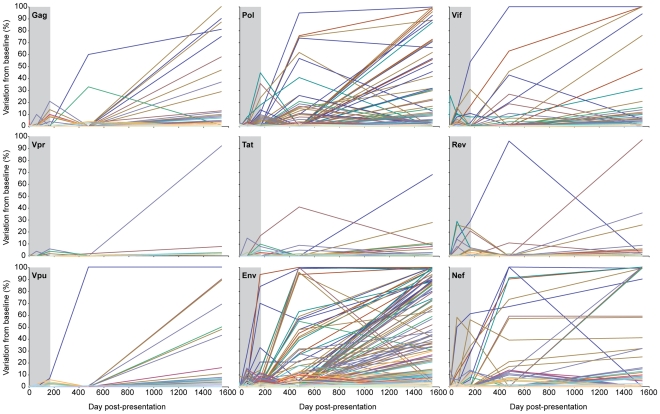
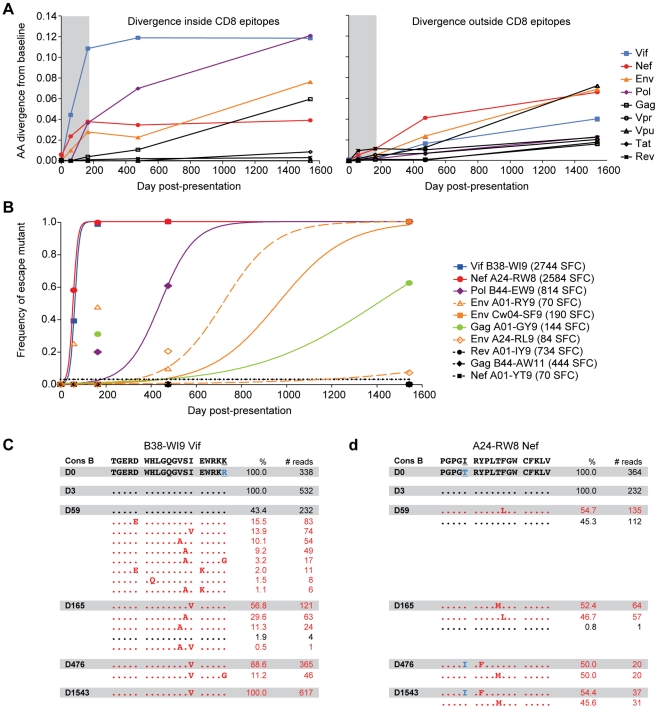
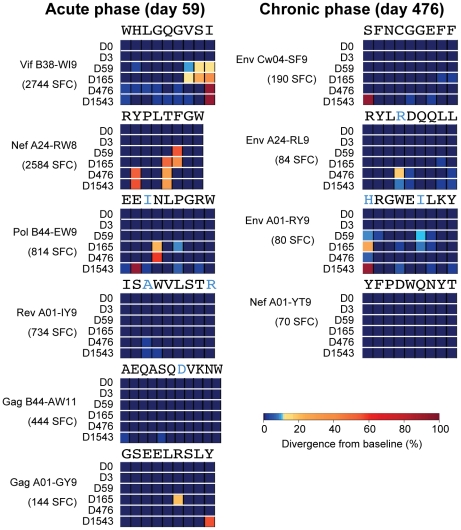
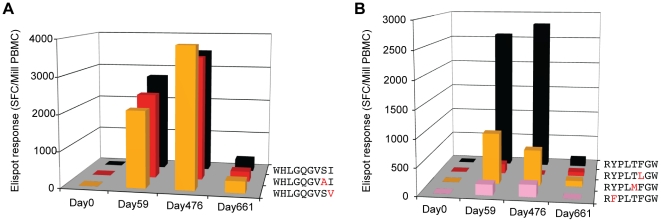
Deep sequencing technologies have the potential to transform the study of highly variable viral pathogens by providing a rapid and cost-effective approach to sensitively characterize rapidly evolving viral quasispecies. Here, we report on a high-throughput whole HIV-1 genome deep sequencing platform that combines 454 pyrosequencing with novel assembly and variant detection algorithms. In one subject we combined these genetic data with detailed immunological analyses to comprehensively evaluate viral evolution and immune escape during the acute phase of HIV-1 infection. The majority of early, low frequency mutations represented viral adaptation to host CD8+ T cell responses, evidence of strong immune selection pressure occurring during the early decline from peak viremia. CD8+ T cell responses capable of recognizing these low frequency escape variants coincided with the selection and evolution of more effective secondary HLA-anchor escape mutations. Frequent, and in some cases rapid, reversion of transmitted mutations was also observed across the viral genome. When located within restricted CD8 epitopes these low frequency reverting mutations were sufficient to prime de novo responses to these epitopes, again illustrating the capacity of the immune response to recognize and respond to low frequency variants. More importantly, rapid viral escape from the most immunodominant CD8+ T cell responses coincided with plateauing of the initial viral load decline in this subject, suggestive of a potential link between maintenance of effective, dominant CD8 responses and the degree of early viremia reduction. We conclude that the early control of HIV-1 replication by immunodominant CD8+ T cell responses may be substantially influenced by rapid, low frequency viral adaptations not detected by conventional sequencing approaches, which warrants further investigation. These data support the critical need for vaccine-induced CD8+ T cell responses to target more highly constrained regions of the virus in order to ensure the maintenance of immunodominant CD8 responses and the sustained decline of early viremia.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing.PLoS One. 2010 Aug 20;5(8):e12303. doi: 10.1371/journal.pone.0012303. PLoS One. 2010. PMID: 20808830 Free PMC article.
-
Antisense-Derived HIV-1 Cryptic Epitopes Are Not Major Drivers of Viral Evolution during the Acute Phase of Infection.J Virol. 2018 Sep 12;92(19):e00711-18. doi: 10.1128/JVI.00711-18. Print 2018 Oct 1. J Virol. 2018. PMID: 30021907 Free PMC article.
-
Two Distinct Mechanisms Leading to Loss of Virological Control in the Rare Group of Antiretroviral Therapy-Naive, Transiently Aviremic Children Living with HIV.J Virol. 2022 Jan 26;96(2):e0153521. doi: 10.1128/JVI.01535-21. Epub 2021 Nov 10. J Virol. 2022. PMID: 34757843 Free PMC article.
-
Role of CD8+ T Cells in the Selection of HIV-1 Immune Escape Mutations.Viral Immunol. 2017 Jan/Feb;30(1):3-12. doi: 10.1089/vim.2016.0095. Epub 2016 Nov 2. Viral Immunol. 2017. PMID: 27805477 Review.
-
Immunodominance of HIV-1-specific CD8(+) T-cell responses in acute HIV-1 infection: at the crossroads of viral and host genetics.Trends Immunol. 2005 Mar;26(3):166-71. doi: 10.1016/j.it.2005.01.003. Trends Immunol. 2005. PMID: 15745859 Review.
Cited by
-
HIV-1 clade B pol evolution following primary infection.PLoS One. 2013 Jun 28;8(6):e68188. doi: 10.1371/journal.pone.0068188. Print 2013. PLoS One. 2013. PMID: 23840830 Free PMC article.
-
Disease progression despite protective HLA expression in an HIV-infected transmission pair.Retrovirology. 2015 Jun 30;12:55. doi: 10.1186/s12977-015-0179-z. Retrovirology. 2015. PMID: 26123575 Free PMC article.
-
Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data.Front Microbiol. 2012 Sep 11;3:329. doi: 10.3389/fmicb.2012.00329. eCollection 2012. Front Microbiol. 2012. PMID: 22973268 Free PMC article.
-
Recent advances in molecular medicine techniques for the diagnosis, prevention, and control of infectious diseases.Eur J Clin Microbiol Infect Dis. 2013 Jun;32(6):723-8. doi: 10.1007/s10096-013-1813-0. Epub 2013 Jan 22. Eur J Clin Microbiol Infect Dis. 2013. PMID: 23339016 Free PMC article. Review.
-
Distinct evolutionary pressures underlie diversity in simian immunodeficiency virus and human immunodeficiency virus lineages.J Virol. 2012 Dec;86(24):13217-31. doi: 10.1128/JVI.01862-12. Epub 2012 Oct 10. J Virol. 2012. PMID: 23055550 Free PMC article.
References
-
- Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407:386–390. - PubMed
-
- Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. - PubMed
-
- Kwiatkowski D, Marsh K. Development of a malaria vaccine. Lancet. 1997;350:1696–1701. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
