

Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth
- PMID: 22393126
- PMCID: PMC3302235
- DOI: 10.1084/jem.20111424
Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth
Abstract
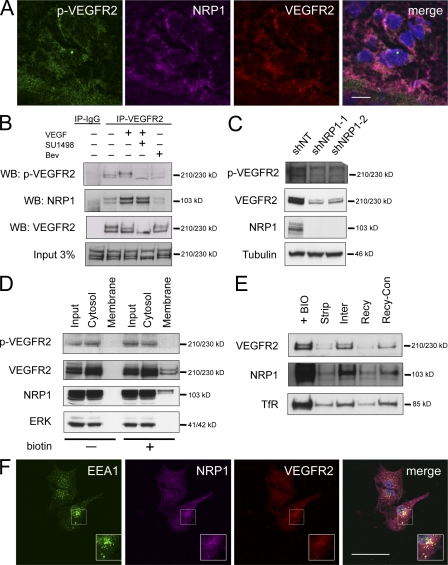
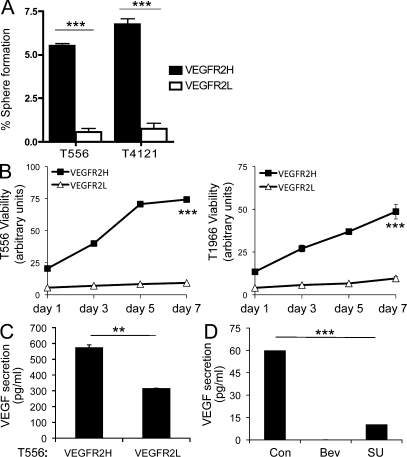
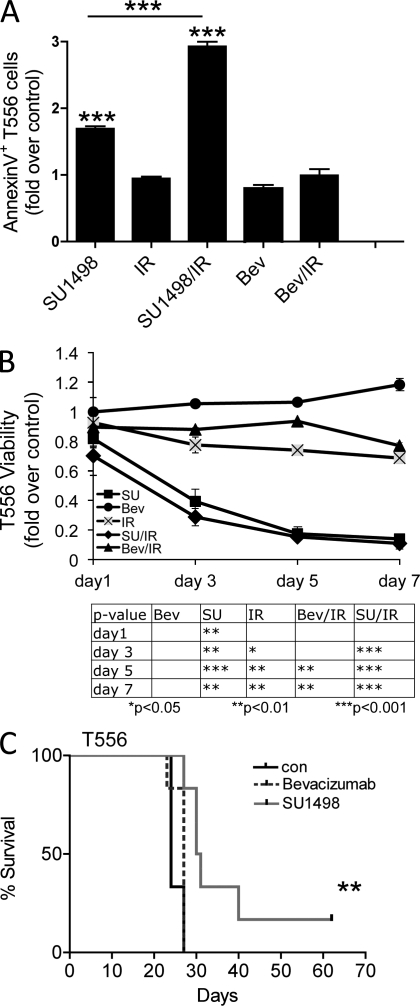
Although vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2) is traditionally regarded as an endothelial cell protein, evidence suggests that VEGFRs may be expressed by cancer cells. Glioblastoma multiforme (GBM) is a lethal cancer characterized by florid vascularization and aberrantly elevated VEGF. Antiangiogenic therapy with the humanized VEGF antibody bevacizumab reduces GBM tumor growth; however, the clinical benefits are transient and invariably followed by tumor recurrence. In this study, we show that VEGFR2 is preferentially expressed on the cell surface of the CD133(+) human glioma stem-like cells (GSCs), whose viability, self-renewal, and tumorigenicity rely, at least in part, on signaling through the VEGF-VEGFR2-Neuropilin-1 (NRP1) axis. We find that the limited impact of bevacizumab-mediated VEGF blockage may reflect ongoing autocrine signaling through VEGF-VEGFR2-NRP1, which is associated with VEGFR2-NRP1 recycling and a pool of active VEGFR2 within a cytosolic compartment of a subset of human GBM cells. Whereas bevacizumab failed to inhibit prosurvival effects of VEGFR2-mediated signaling, GSC viability under unperturbed or radiation-evoked stress conditions was attenuated by direct inhibition of VEGFR2 tyrosine kinase activity and/or shRNA-mediated knockdown of VEGFR2 or NRP1. We propose that direct inhibition of VEGFR2 kinase may block the highly dynamic VEGF-VEGFR2-NRP1 pathway and inspire a GBM treatment strategy to complement the currently prevalent ligand neutralization approach.
Figures



Similar articles
-
The effect of bevacizumab on human malignant melanoma cells with functional VEGF/VEGFR2 autocrine and intracrine signaling loops.Neoplasia. 2012 Jul;14(7):612-23. doi: 10.1593/neo.11948. Neoplasia. 2012. PMID: 22904678 Free PMC article.
-
VEGF-C sustains VEGFR2 activation under bevacizumab therapy and promotes glioblastoma maintenance.Neuro Oncol. 2018 Oct 9;20(11):1462-1474. doi: 10.1093/neuonc/noy103. Neuro Oncol. 2018. PMID: 29939339 Free PMC article.
-
Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo.Neuro Oncol. 2016 Sep;18(9):1242-52. doi: 10.1093/neuonc/now043. Epub 2016 Mar 23. Neuro Oncol. 2016. PMID: 27009237 Free PMC article.
-
Anti-VEGF/VEGFR2 Monoclonal Antibodies and their Combinations with PD-1/PD-L1 Inhibitors in Clinic.Curr Cancer Drug Targets. 2020;20(1):3-18. doi: 10.2174/1568009619666191114110359. Curr Cancer Drug Targets. 2020. PMID: 31729943 Review.
-
VEGF-A121a binding to Neuropilins - A concept revisited.Cell Adh Migr. 2018 May 4;12(3):204-214. doi: 10.1080/19336918.2017.1372878. Epub 2017 Nov 2. Cell Adh Migr. 2018. PMID: 29095088 Free PMC article. Review.
Cited by
-
GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation.EMBO Mol Med. 2013 Apr;5(4):488-508. doi: 10.1002/emmm.201202078. Epub 2013 Feb 21. EMBO Mol Med. 2013. PMID: 23436775 Free PMC article.
-
Serpin family A member 1 is an oncogene in glioma and its translation is enhanced by NAD(P)H quinone dehydrogenase 1 through RNA-binding activity.Open Med (Wars). 2022 Oct 20;17(1):1645-1654. doi: 10.1515/med-2022-0572. eCollection 2022. Open Med (Wars). 2022. PMID: 36349191 Free PMC article.
-
Intratumor heterogeneity, microenvironment, and mechanisms of drug resistance in glioma recurrence and evolution.Front Med. 2021 Aug;15(4):551-561. doi: 10.1007/s11684-020-0760-2. Epub 2021 Apr 24. Front Med. 2021. PMID: 33893983 Review.
-
CRISPR-mediated knockout of VEGFR2/KDR inhibits cell growth in a squamous thyroid cancer cell line.FEBS Open Bio. 2022 May;12(5):993-1005. doi: 10.1002/2211-5463.13399. Epub 2022 Apr 8. FEBS Open Bio. 2022. PMID: 35313079 Free PMC article.
-
Hedgehog signaling regulates the development and treatment of glioblastoma.Oncol Lett. 2022 Jul 5;24(3):294. doi: 10.3892/ol.2022.13414. eCollection 2022 Sep. Oncol Lett. 2022. PMID: 35949611 Free PMC article. Review.
References
-
- Beier D., Hau P., Proescholdt M., Lohmeier A., Wischhusen J., Oefner P.J., Aigner L., Brawanski A., Bogdahn U., Beier C.P. 2007. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 67:4010–4015 10.1158/0008-5472.CAN-06-4180 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
