

A DNA hypermethylation module for the stem/progenitor cell signature of cancer
- PMID: 22391556
- PMCID: PMC3337430
- DOI: 10.1101/gr.131169.111
A DNA hypermethylation module for the stem/progenitor cell signature of cancer
Abstract
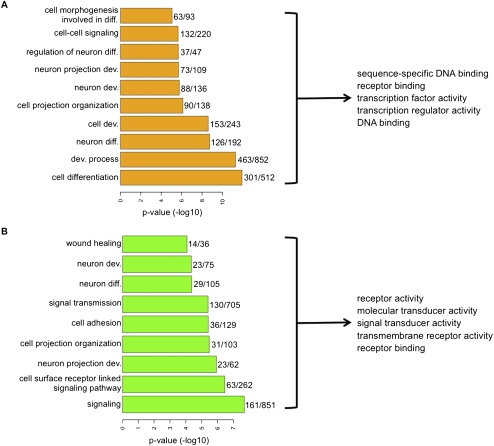
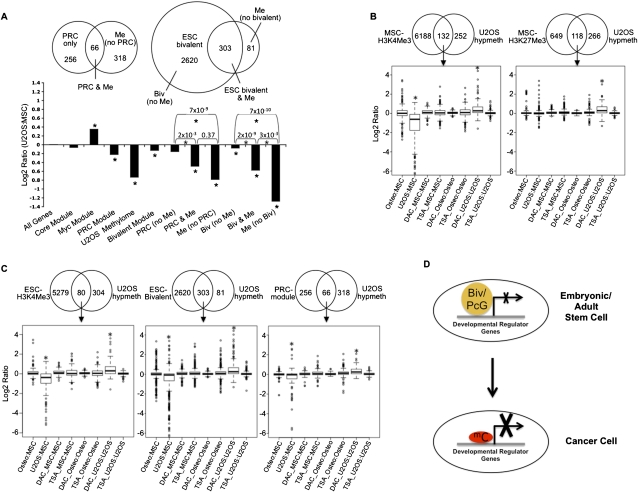
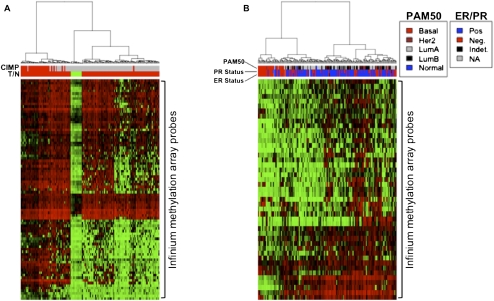
Many DNA-hypermethylated cancer genes are occupied by the Polycomb (PcG) repressor complex in embryonic stem cells (ESCs). Their prevalence in the full spectrum of cancers, the exact context of chromatin involved, and their status in adult cell renewal systems are unknown. Using a genome-wide analysis, we demonstrate that ~75% of hypermethylated genes are marked by PcG in the context of bivalent chromatin in both ESCs and adult stem/progenitor cells. A large number of these genes are key developmental regulators, and a subset, which we call the "DNA hypermethylation module," comprises a portion of the PcG target genes that are down-regulated in cancer. Genes with bivalent chromatin have a low, poised gene transcription state that has been shown to maintain stemness and self-renewal in normal stem cells. However, when DNA-hypermethylated in tumors, we find that these genes are further repressed. We also show that the methylation status of these genes can cluster important subtypes of colon and breast cancers. By evaluating the subsets of genes that are methylated in different cancers with consideration of their chromatin status in ESCs, we provide evidence that DNA hypermethylation preferentially targets the subset of PcG genes that are developmental regulators, and this may contribute to the stem-like state of cancer. Additionally, the capacity for global methylation profiling to cluster tumors by phenotype may have important implications for further refining tumor behavior patterns that may ultimately aid therapeutic interventions.
Figures
Similar articles
-
PcG proteins, DNA methylation, and gene repression by chromatin looping.PLoS Biol. 2008 Dec 2;6(12):2911-27. doi: 10.1371/journal.pbio.0060306. PLoS Biol. 2008. PMID: 19053175 Free PMC article.
-
Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells.Cancer Res. 2008 Jul 15;68(14):5753-9. doi: 10.1158/0008-5472.CAN-08-0700. Cancer Res. 2008. PMID: 18632628 Free PMC article.
-
An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research.Oncotarget. 2017 Jan 17;8(3):4110-4124. doi: 10.18632/oncotarget.13746. Oncotarget. 2017. PMID: 27926531 Free PMC article.
-
Role of the polycomb group proteins in hematopoietic stem cells.Dev Growth Differ. 2010 Aug;52(6):505-16. doi: 10.1111/j.1440-169X.2010.01191.x. Dev Growth Differ. 2010. PMID: 20646023 Review.
-
Identification of driver and passenger DNA methylation in cancer by epigenomic analysis.Adv Genet. 2010;70:277-308. doi: 10.1016/B978-0-12-380866-0.60010-1. Adv Genet. 2010. PMID: 20920752 Free PMC article. Review.
Cited by
-
Maintaining cell identity: PRC2-mediated regulation of transcription and cancer.Nat Rev Cancer. 2016 Dec;16(12):803-810. doi: 10.1038/nrc.2016.83. Epub 2016 Sep 23. Nat Rev Cancer. 2016. PMID: 27658528 Review.
-
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch.Sci Adv. 2024 Aug 30;10(35):eadp0975. doi: 10.1126/sciadv.adp0975. Epub 2024 Aug 28. Sci Adv. 2024. PMID: 39196936 Free PMC article.
-
Real-time methylomic aberrations during initiation and progression of induced human mammary epithelial cell tumorigenesis.Epigenomics. 2013 Apr;5(2):155-65. doi: 10.2217/epi.13.6. Epigenomics. 2013. PMID: 23566093 Free PMC article.
-
Signals that regulate the oncogenic fate of neural stem cells and progenitors.Exp Neurol. 2014 Oct;260:56-68. doi: 10.1016/j.expneurol.2013.01.027. Epub 2013 Jan 31. Exp Neurol. 2014. PMID: 23376224 Free PMC article. Review.
-
Epigenetic mechanisms of tumorigenicity manifesting in stem cells.Oncogene. 2015 Apr 30;34(18):2288-96. doi: 10.1038/onc.2014.172. Epub 2014 Jun 16. Oncogene. 2015. PMID: 24931168 Free PMC article. Review.
References
-
- Baylin S, Bestor TH 2002. Altered methylation patterns in cancer cell genomes: Cause or consequence? Cancer Cell 1: 299–305 - PubMed
-
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300
-
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, et al. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181 - PubMed
-
- Bibikova M, Fan JB 2009. GoldenGate assay for DNA methylation profiling. Methods Mol Biol 507: 149–163 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous