

Order out of disorder: working cycle of an intrinsically unfolded chaperone
- PMID: 22385960
- PMCID: PMC3376891
- DOI: 10.1016/j.cell.2012.01.045
Order out of disorder: working cycle of an intrinsically unfolded chaperone
Abstract
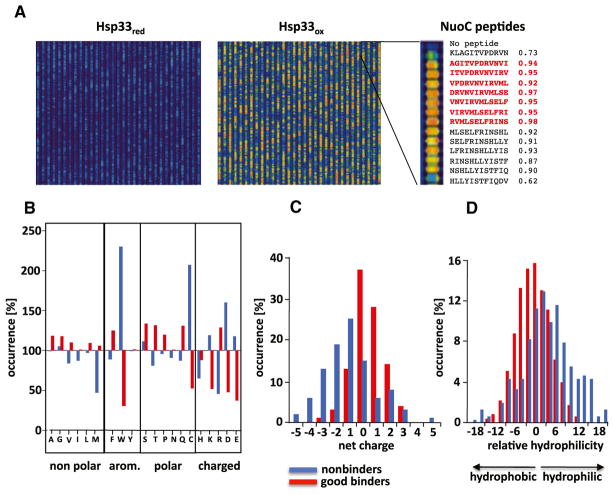
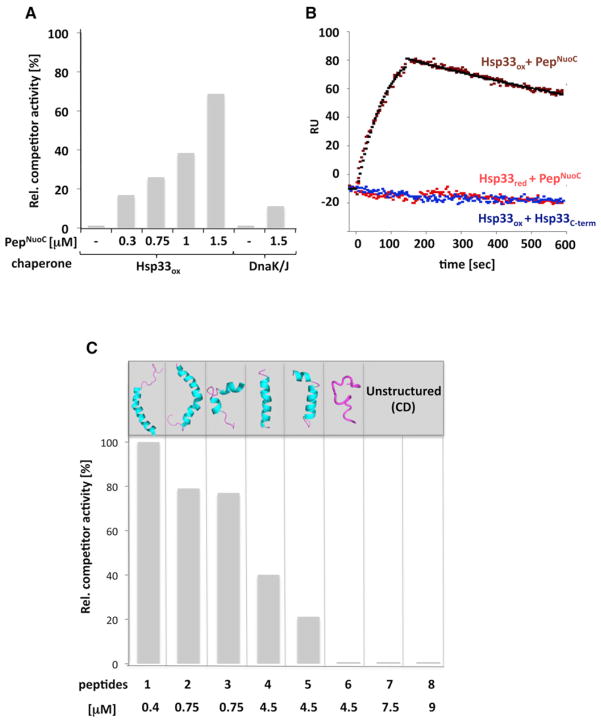
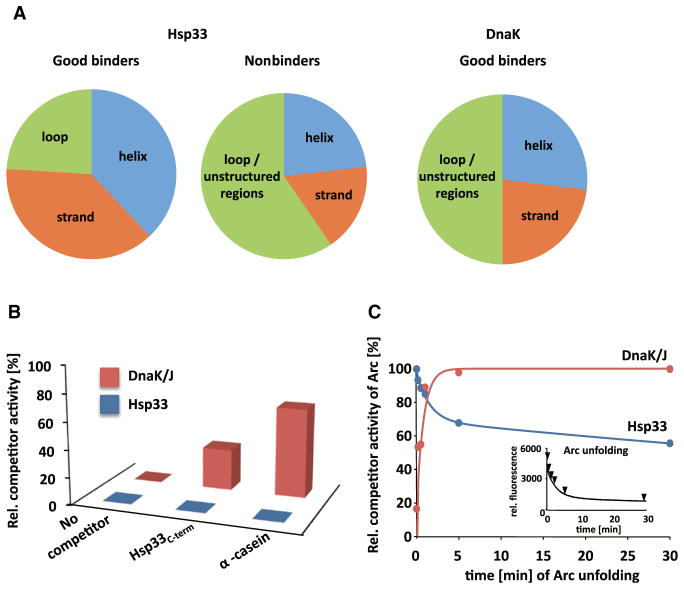
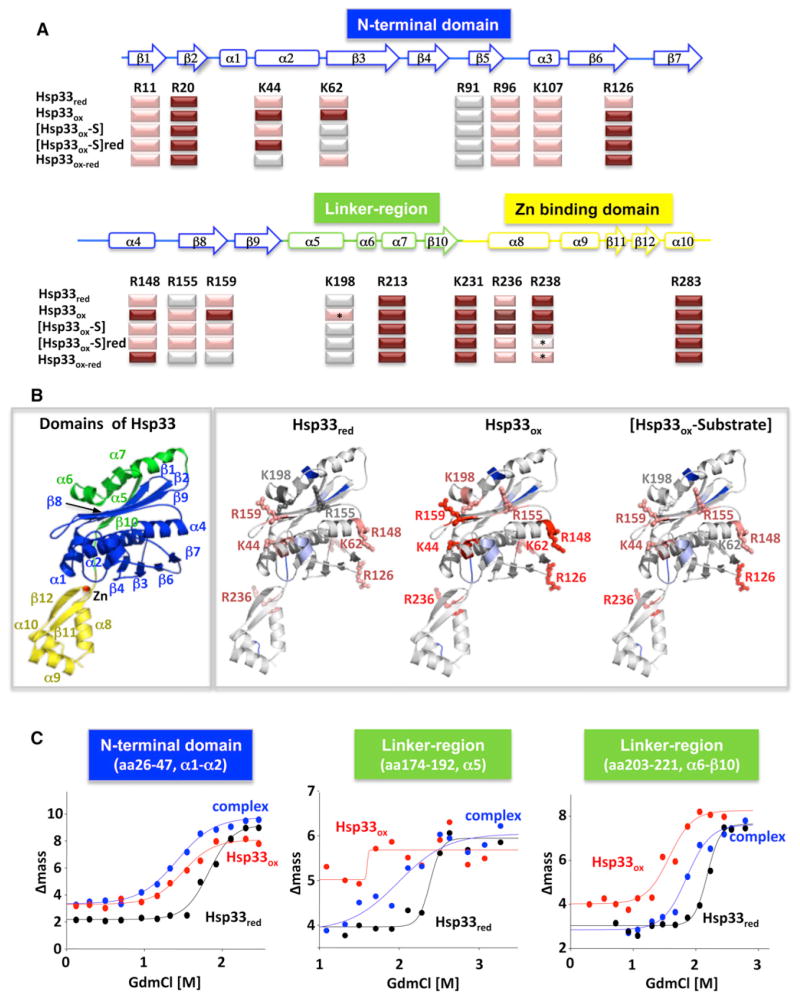
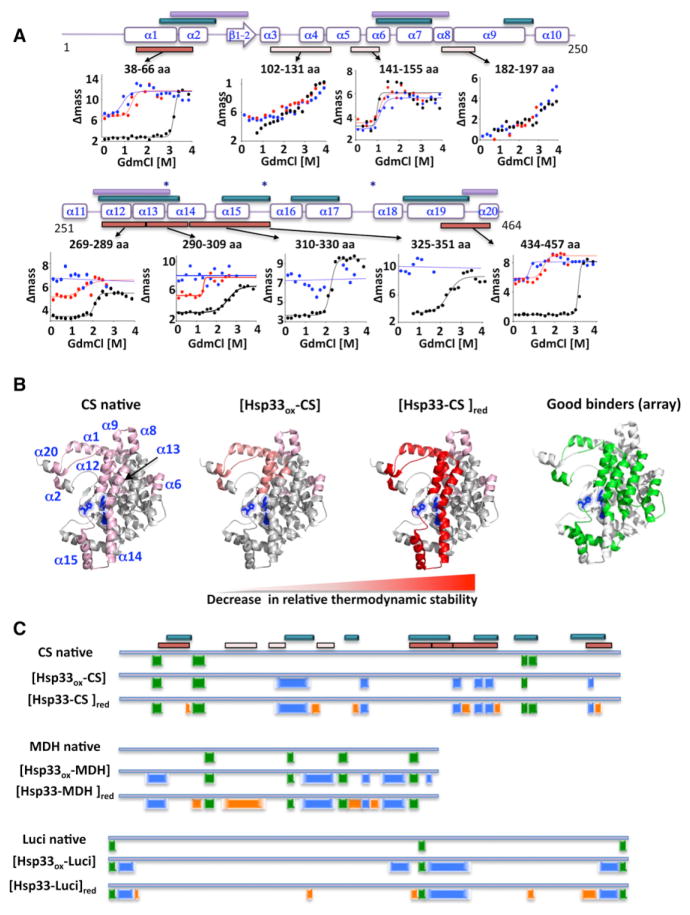
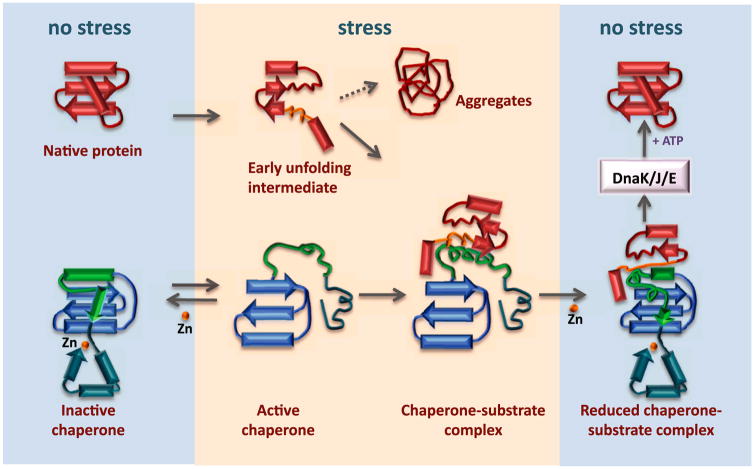
The redox-regulated chaperone Hsp33 protects organisms against oxidative stress that leads to protein unfolding. Activation of Hsp33 is triggered by the oxidative unfolding of its own redox-sensor domain, making Hsp33 a member of a recently discovered class of chaperones that require partial unfolding for full chaperone activity. Here we address the long-standing question of how chaperones recognize client proteins. We show that Hsp33 uses its own intrinsically disordered regions to discriminate between unfolded and partially structured folding intermediates. Binding to secondary structure elements in client proteins stabilizes Hsp33's intrinsically disordered regions, and this stabilization appears to mediate Hsp33's high affinity for structured folding intermediates. Return to nonstress conditions reduces Hsp33's disulfide bonds, which then significantly destabilizes the bound client proteins and in doing so converts them into less-structured, folding-competent client proteins of ATP-dependent foldases. We propose a model in which energy-independent chaperones use internal order-to-disorder transitions to control substrate binding and release.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Comment in
-
The unfolding story of a redox chaperone.Cell. 2012 Mar 2;148(5):843-4. doi: 10.1016/j.cell.2012.02.029. Cell. 2012. PMID: 22385952
Similar articles
-
A Role of Metastable Regions and Their Connectivity in the Inactivation of a Redox-Regulated Chaperone and Its Inter-Chaperone Crosstalk.Antioxid Redox Signal. 2017 Nov 20;27(15):1252-1267. doi: 10.1089/ars.2016.6900. Epub 2017 Apr 10. Antioxid Redox Signal. 2017. PMID: 28394178
-
Protein unfolding as a switch from self-recognition to high-affinity client binding.Nat Commun. 2016 Jan 20;7:10357. doi: 10.1038/ncomms10357. Nat Commun. 2016. PMID: 26787517 Free PMC article.
-
Activation of the redox-regulated molecular chaperone Hsp33--a two-step mechanism.Structure. 2001 May 9;9(5):377-87. doi: 10.1016/s0969-2126(01)00599-8. Structure. 2001. PMID: 11377198
-
Redox-regulated molecular chaperones.Cell Mol Life Sci. 2002 Oct;59(10):1624-31. doi: 10.1007/pl00012489. Cell Mol Life Sci. 2002. PMID: 12475172 Free PMC article. Review.
-
Interferon-gamma is a target for binding and folding by both Escherichia coli chaperone model systems GroEL/GroES and DnaK/DnaJ/GrpE.Biochimie. 1998 Aug-Sep;80(8-9):729-37. doi: 10.1016/s0300-9084(99)80026-1. Biochimie. 1998. PMID: 9865495 Review.
Cited by
-
Thiol-based redox switches.Biochim Biophys Acta. 2014 Aug;1844(8):1335-43. doi: 10.1016/j.bbapap.2014.03.007. Epub 2014 Mar 19. Biochim Biophys Acta. 2014. PMID: 24657586 Free PMC article. Review.
-
Bacterial Defense Systems against the Neutrophilic Oxidant Hypochlorous Acid.Infect Immun. 2020 Jun 22;88(7):e00964-19. doi: 10.1128/IAI.00964-19. Print 2020 Jun 22. Infect Immun. 2020. PMID: 32152198 Free PMC article. Review.
-
Protein Folding and Mechanisms of Proteostasis.Int J Mol Sci. 2015 Jul 28;16(8):17193-230. doi: 10.3390/ijms160817193. Int J Mol Sci. 2015. PMID: 26225966 Free PMC article. Review.
-
Forces Driving Chaperone Action.Cell. 2016 Jul 14;166(2):369-379. doi: 10.1016/j.cell.2016.05.054. Epub 2016 Jun 9. Cell. 2016. PMID: 27293188 Free PMC article.
-
Small heat shock protein IbpB acts as a robust chaperone in living cells by hierarchically activating its multi-type substrate-binding residues.J Biol Chem. 2013 Apr 26;288(17):11897-906. doi: 10.1074/jbc.M113.450437. Epub 2013 Mar 13. J Biol Chem. 2013. PMID: 23486475 Free PMC article.
References
-
- Bowie JU, Sauer RT. Equilibrium dissociation and unfolding of the Arc repressor dimer. Biochemistry. 1989;28:7139–7143. - PubMed
-
- Chen Z, Kurt N, Rajagopalan S, Cavagnero S. Secondary structure mapping of DnaK-bound protein fragments: chain helicity and local helix unwinding at the binding site. Biochemistry. 2006;45:12325–12333. - PubMed
-
- Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. - PubMed
-
- Gaspar AM, Appavou MS, Busch S, Unruh T, Doster W. Dynamics of well-folded and natively disordered proteins in solution: a time-of-flight neutron scattering study. Eur Biophys J. 2008;37:573–582. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
