

CXCL13 activation of c-Myc induces RANK ligand expression in stromal/preosteoblast cells in the oral squamous cell carcinoma tumor-bone microenvironment
- PMID: 22330139
- PMCID: PMC3355224
- DOI: 10.1038/onc.2012.24
CXCL13 activation of c-Myc induces RANK ligand expression in stromal/preosteoblast cells in the oral squamous cell carcinoma tumor-bone microenvironment
Abstract
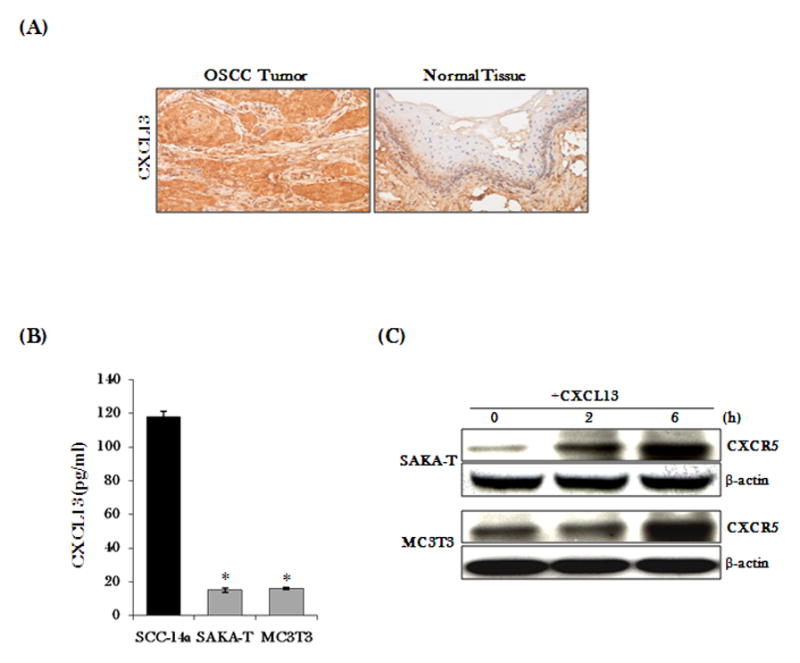
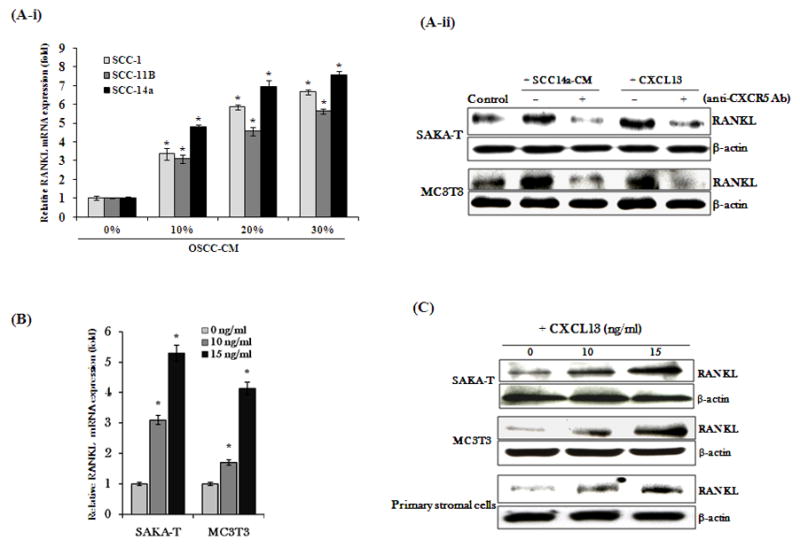
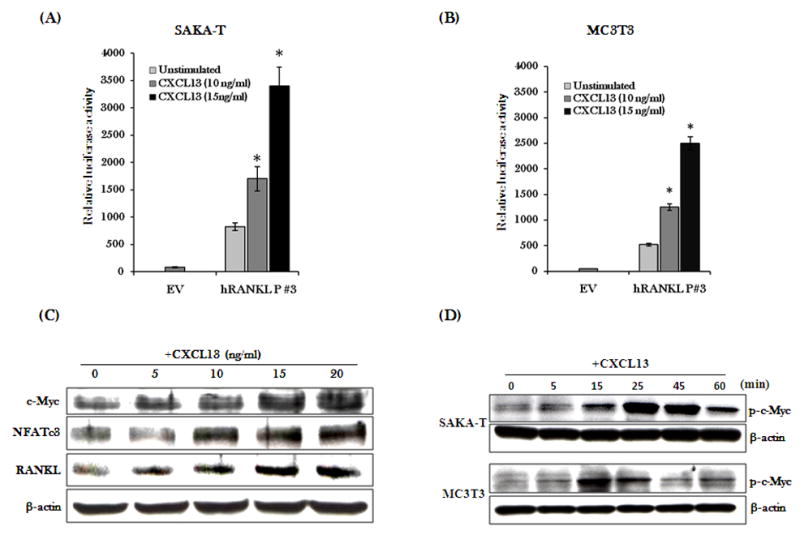
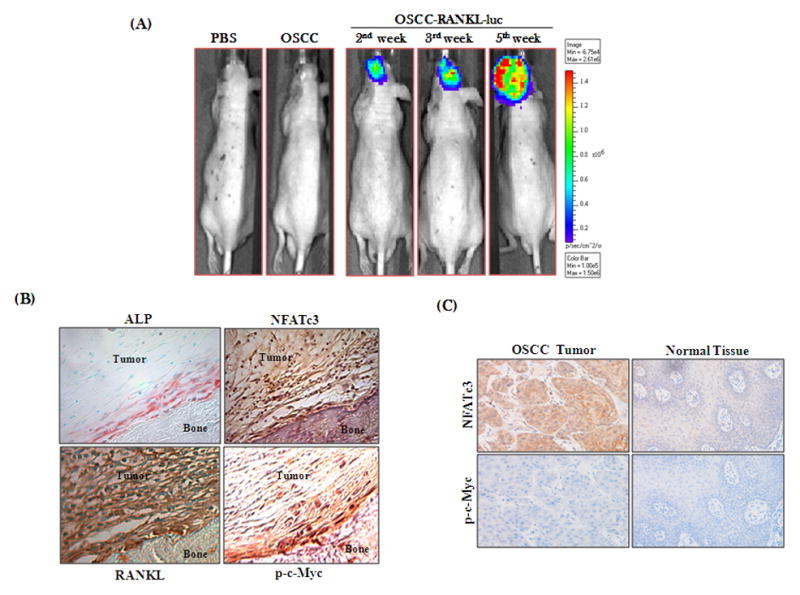
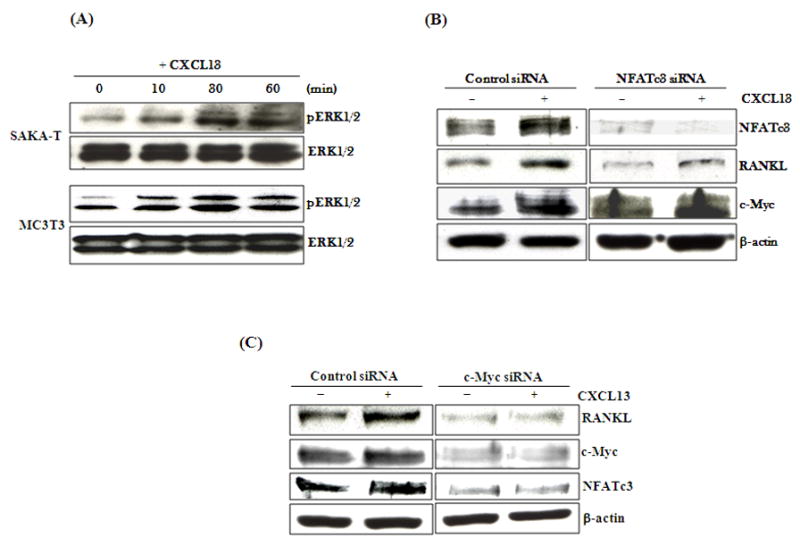
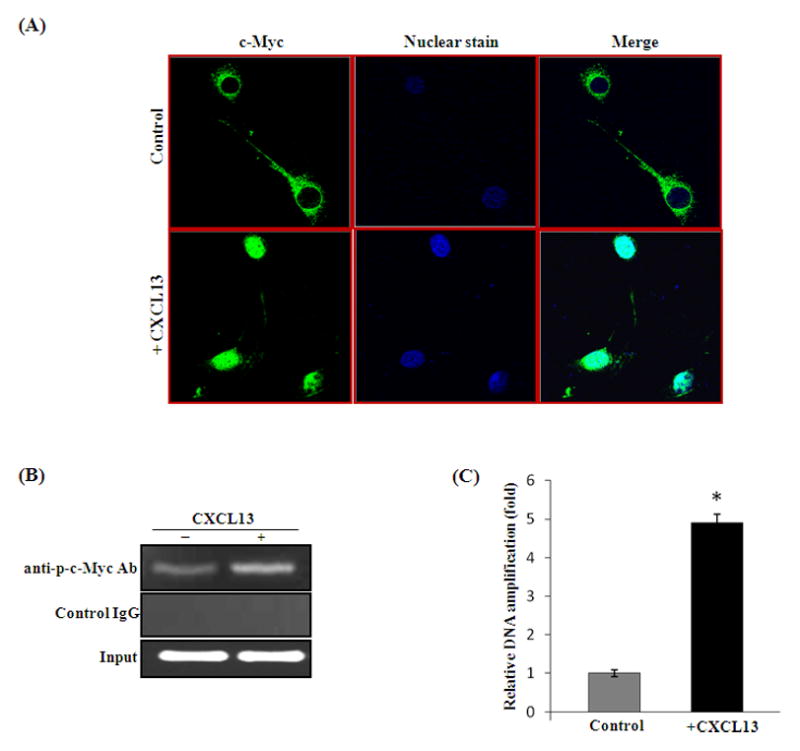
CXC chemokine ligand-13 (CXCL13) has been implicated in oral squamous cell carcinoma (OSCC) tumor progression and osteolysis. The tumor necrosis factor family member RANKL (receptor activator of NF-κB ligand), a critical bone resorbing osteoclastogenic factor, has an important role in cancer invasion of bone/osteolysis. Here, we show high-level expression of CXCL13 in primary human OSCC tumor specimens; however, human bone marrow-derived stromal (SAKA-T) and murine preosteoblast (MC3T3-E1) cells produce at very low level. Recombinant CXCL13 (0-15 ng/ml) dose dependently induced CXCR5 expression in SAKA-T and MC3T3-E1 cells. Conditioned media obtained from OSCC cell lines increased the RANKL expression and an antibody against the CXCL13 specific receptor, CXCR5 markedly decreased RANKL expression in these cells. Furthermore, CXCL13 increased hRANKL-Luc promoter activity. Superarray screening identified c-Myc and NFATc3 transcription factors upregulated in CXCL13-stimulated SAKA-T cells. Immunohistochemical analysis of OSCC tumors that developed in athymic mice demonstrated RANKL and NFATc3 expression in tumor and osteoblast cells, however, showed p-c-Myc expression specific to osteoblastic cells at the tumor-bone interface. We further identified NFATc3 expression, but not c-Myc activation in primary human OSCC tumor specimens compared with adjacent normal tissue. Also, CXCL13 significantly increased p-ERK1/2 in SAKA-T and MC3T3-E1 cells. siRNA suppression of c-Myc expression markedly decreased CXCL13-induced RANKL and NFATc3 expression in preosteoblast cells. Chromatin-immuno precipitation assay confirmed p-c-Myc binding to the hRANKL promoter region. In summary, c-Myc activation through CXCL13-CXCR5 signaling axis stimulates RANKL expression in stromal/preosteoblast cells. Thus, our results implicate CXCL13 as a potential therapeutic target to prevent OSCC invasion of bone/osteolysis.
Conflict of interest statement
The authors declare no conflict of interest with this work.
Figures
Similar articles
-
A novel function of CXCL13 to stimulate RANK ligand expression in oral squamous cell carcinoma cells.Mol Cancer Res. 2009 Aug;7(8):1399-407. doi: 10.1158/1541-7786.MCR-08-0589. Epub 2009 Aug 11. Mol Cancer Res. 2009. PMID: 19671684
-
Role of CXC chemokine ligand 13 in oral squamous cell carcinoma associated osteolysis in athymic mice.Int J Cancer. 2010 May 15;126(10):2319-29. doi: 10.1002/ijc.24920. Int J Cancer. 2010. PMID: 19816883 Free PMC article.
-
STAT-6 mediates TRAIL induced RANK ligand expression in stromal/preosteoblast cells.Bone. 2015 Feb;71:137-44. doi: 10.1016/j.bone.2014.10.016. Epub 2014 Oct 30. Bone. 2015. PMID: 25445452
-
The RANKL/RANK system as a therapeutic target for bone invasion by oral squamous cell carcinoma (Review).Int J Oncol. 2013 Mar;42(3):803-9. doi: 10.3892/ijo.2013.1794. Epub 2013 Jan 23. Int J Oncol. 2013. PMID: 23354319 Review.
-
CXCL13/CXCR5 signaling axis in cancer.Life Sci. 2019 Jun 15;227:175-186. doi: 10.1016/j.lfs.2019.04.053. Epub 2019 Apr 23. Life Sci. 2019. PMID: 31026453 Review.
Cited by
-
Unveiling the power of flavonoids: A dynamic exploration of their impact on cancer through matrix metalloproteinases regulation.Biomedicine (Taipei). 2024 Jun 1;14(2):12-28. doi: 10.37796/2211-8039.1447. eCollection 2024. Biomedicine (Taipei). 2024. PMID: 38939095 Free PMC article. Review.
-
Effect of CXCR5-Positive Cell Infiltration on the Immune Contexture and Patient Prognosis in Head and Neck Squamous Cell Carcinoma.Onco Targets Ther. 2020 Jun 22;13:5869-5877. doi: 10.2147/OTT.S248958. eCollection 2020. Onco Targets Ther. 2020. PMID: 32606797 Free PMC article.
-
Transcriptomic analysis identifies differences in gene expression in actinic keratoses after treatment with imiquimod and between responders and non responders.Sci Rep. 2021 Apr 22;11(1):8775. doi: 10.1038/s41598-021-88424-z. Sci Rep. 2021. PMID: 33888854 Free PMC article.
-
CXCL13 in Cancer and Other Diseases: Biological Functions, Clinical Significance, and Therapeutic Opportunities.Life (Basel). 2021 Nov 23;11(12):1282. doi: 10.3390/life11121282. Life (Basel). 2021. PMID: 34947813 Free PMC article. Review.
-
[Advances in molecular mechanisms of bone invasion by oral cancer].Hua Xi Kou Qiang Yi Xue Za Zhi. 2021 Apr 1;39(2):221-226. doi: 10.7518/hxkq.2021.02.015. Hua Xi Kou Qiang Yi Xue Za Zhi. 2021. PMID: 33834679 Free PMC article. Review. Chinese.
References
-
- Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5:311–316. - PubMed
-
- Funk GF, Karnell LH, Robinson RA, Zhen WK, Trask DK, Hoffman HT. Presentation, treatment, and outcome of oral cavity cancer: a National Cancer Data Base report. Head Neck. 2002;24:165–180. - PubMed
-
- Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14–32. - PubMed
-
- Nomura T, Shibahara T, Katakura A, Matsubara S, Takano N. Establishment of a murine model of bone invasion by oral squamous cell carcinoma. Oral Oncol. 2007;43:257–262. - PubMed
-
- Mishra A, Bharti AC, Varghese P, Saluja D, Das BC. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: Role of high risk human papillomavirus infection. Int J Cancer. 2006;119:2840–2850. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
