

Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury
- PMID: 22307460
- PMCID: PMC3689100
- DOI: 10.1093/eurheartj/ehs011
Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury
Abstract
Aims: Emerging evidence indicates that nuclear receptors play a critical regulatory role in cardiovascular physiology/pathology. Recently, farnesoid-X-receptor (FXR), a member of the metabolic nuclear receptor superfamily, has been demonstrated to be expressed in vascular cells, with important roles in vascular physiology/pathology. However, the potential cardiac function of FXR remains unclear. We investigated the cardiac expression and biological function of FXR.
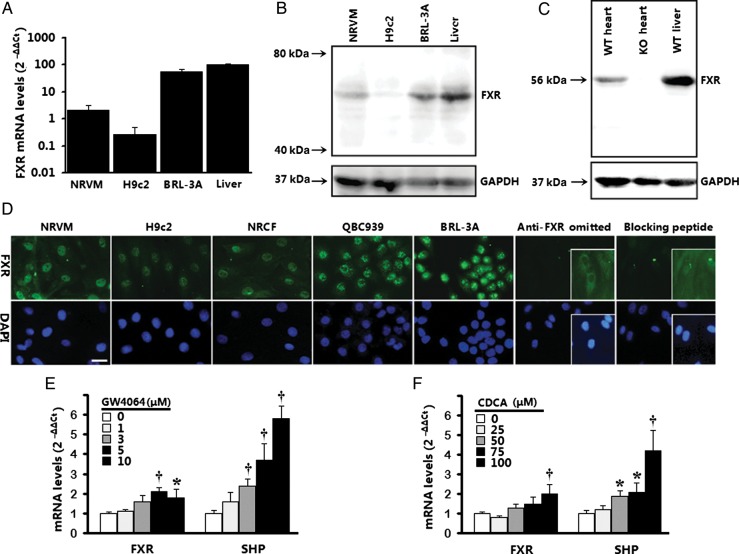
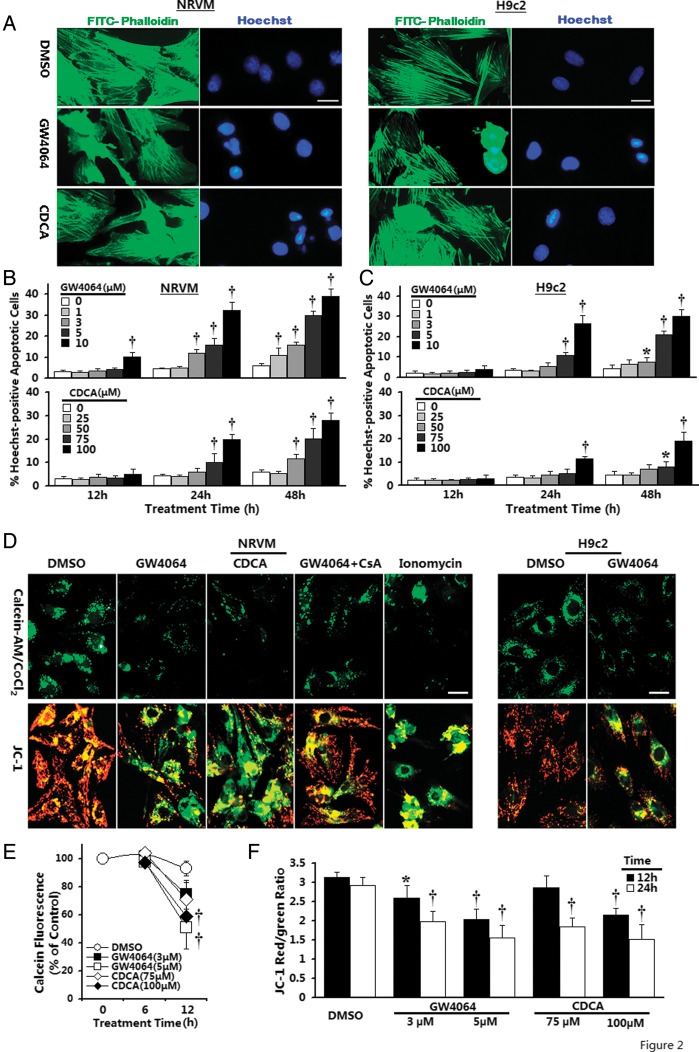
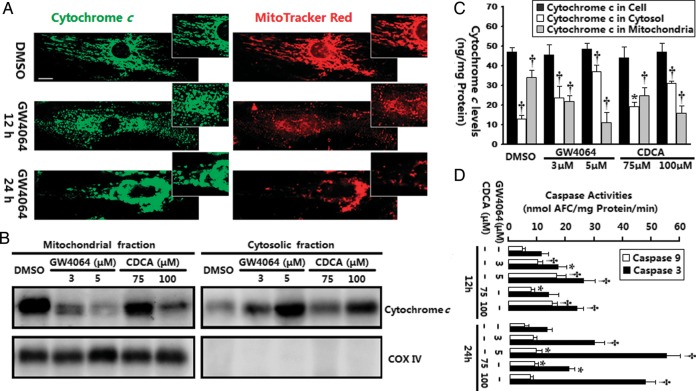
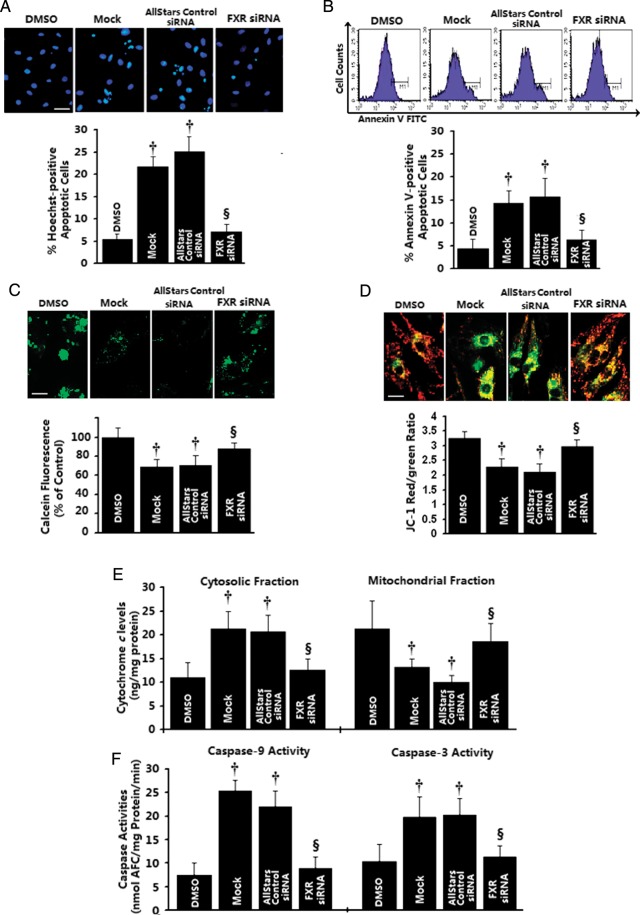
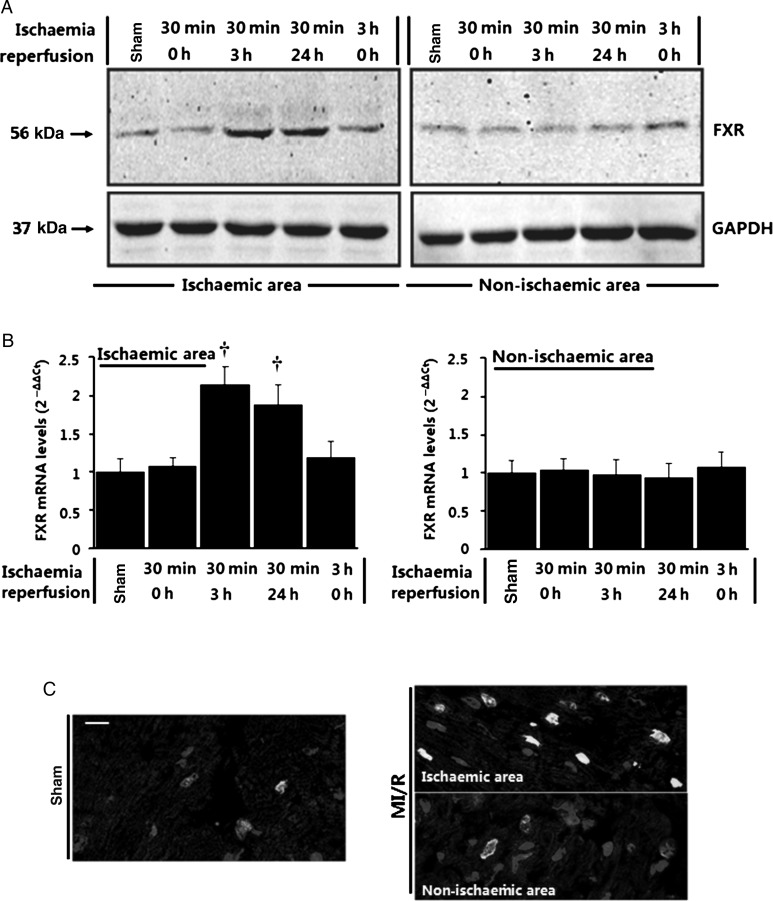
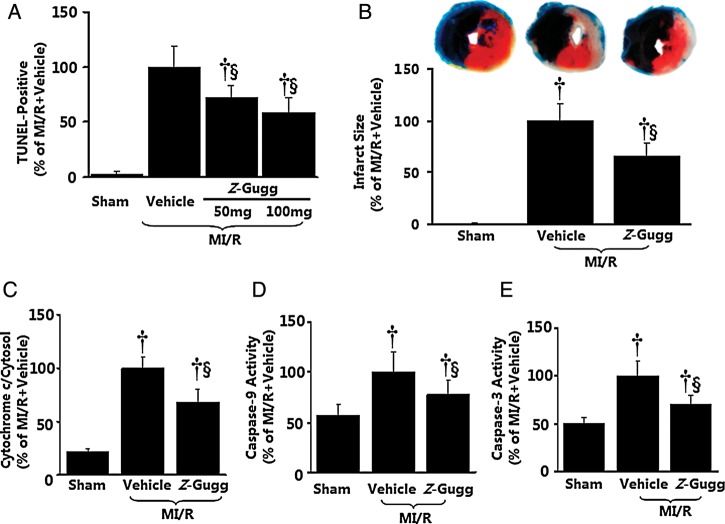
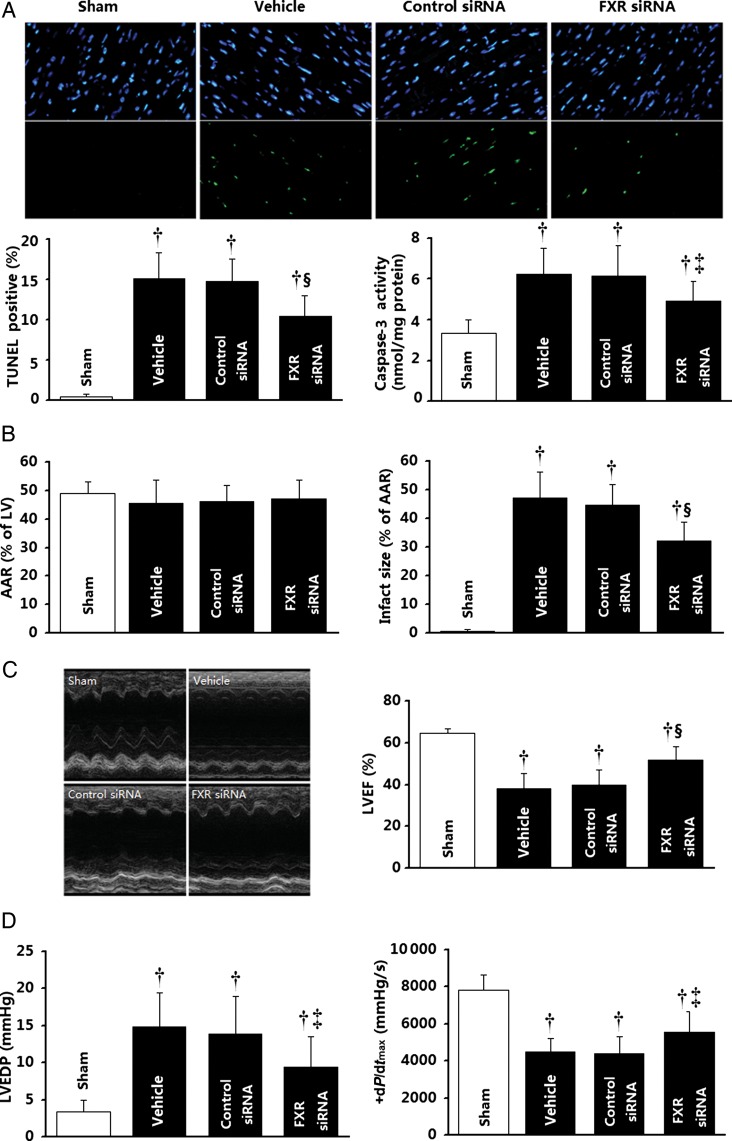
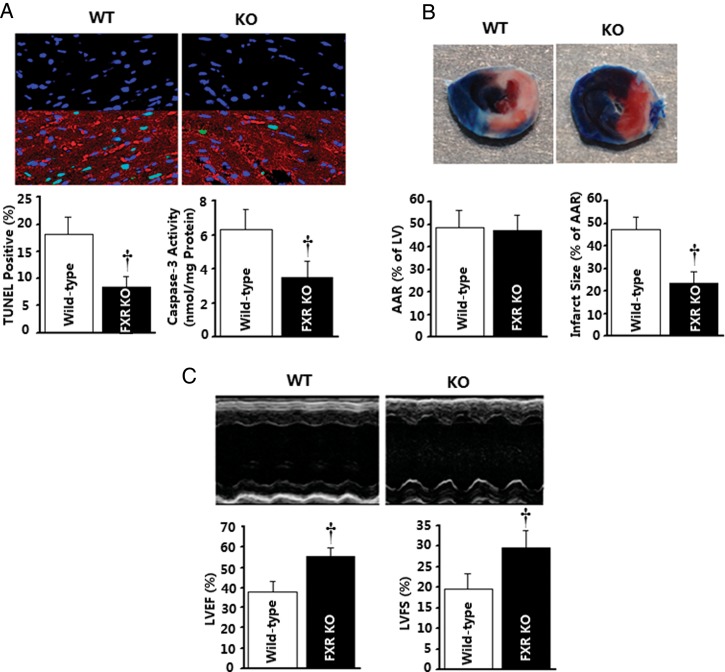
Methods and results: Farnesoid-X-receptor was detected in both isolated neonatal rat cardiac myocytes and fibroblasts. Natural and synthetic FXR agonists upregulated cardiac FXR expression, stimulated myocyte apoptosis, and reduced myocyte viability dose- and time-dependently. Mechanistic studies demonstrated that FXR agonists disrupted mitochondria, characterized by mitochondrial permeability transition pores activation, mitochondrial potential dissipation, cytochrome c release, and both caspase-9 and -3 activation. Such mitochondrial apoptotic responses were abolished by siRNA-mediated silencing of endogenous FXR or pharmacological inhibition of mitochondrial death signalling. Furthermore, low levels of FXR were detected in the adult mouse heart, with significant (∼2.0-fold) upregulation after myocardial ischaemia/reperfusion (MI/R). Pharmacological inhibition or genetic ablation of FXR significantly reduced myocardial apoptosis by 29.0-53.4%, decreased infarct size by 23.4-49.7%, and improved cardiac function in ischaemic/reperfused myocardium.
Conclusion: These results demonstrate that nuclear receptor FXR acts as a novel functional receptor in cardiac tissue, regulates apoptosis in cardiomyocytes, and contributes to MI/R injury.
Keywords: Apoptosis; Myocytes; Nuclear receptors.
Figures
Similar articles
-
[Farnesoid-X-receptor blockade reduces myocardial reperfusion injury in cholesterol-fed apolipoprotein E knockout mice].Zhonghua Xin Xue Guan Bing Za Zhi. 2013 Aug;41(8):642-6. Zhonghua Xin Xue Guan Bing Za Zhi. 2013. PMID: 24225234 Chinese.
-
Adiponectin determines farnesoid X receptor agonism-mediated cardioprotection against post-infarction remodelling and dysfunction.Cardiovasc Res. 2018 Aug 1;114(10):1335-1349. doi: 10.1093/cvr/cvy093. Cardiovasc Res. 2018. PMID: 29668847
-
Panax quinquefolium saponin attenuates cardiomyocyte apoptosis and opening of the mitochondrial permeability transition pore in a rat model of ischemia/reperfusion.Cell Physiol Biochem. 2014;34(4):1413-26. doi: 10.1159/000366347. Epub 2014 Oct 3. Cell Physiol Biochem. 2014. PMID: 25301366
-
Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue.J Bioenerg Biomembr. 2009 Apr;41(2):169-80. doi: 10.1007/s10863-009-9205-y. J Bioenerg Biomembr. 2009. PMID: 19377835 Free PMC article. Review.
-
Targeted mitochondrial drugs for treatment of myocardial ischaemia-reperfusion injury.J Drug Target. 2022 Sep;30(8):833-844. doi: 10.1080/1061186X.2022.2085728. Epub 2022 Jun 28. J Drug Target. 2022. PMID: 35652502 Review.
Cited by
-
Toll-interacting protein contributes to mortality following myocardial infarction through promoting inflammation and apoptosis.Br J Pharmacol. 2015 Jul;172(13):3383-96. doi: 10.1111/bph.13130. Epub 2015 Apr 24. Br J Pharmacol. 2015. PMID: 25765712 Free PMC article.
-
Proteomic Analysis of Effects of Spironolactone in Heart Failure With Preserved Ejection Fraction.Circ Heart Fail. 2022 Sep;15(9):e009693. doi: 10.1161/CIRCHEARTFAILURE.121.009693. Epub 2022 Aug 9. Circ Heart Fail. 2022. PMID: 36126144 Free PMC article. Clinical Trial.
-
Biliary cirrhosis-induced cardiac abnormality in rats: Interaction between Farnesoid-X-activated receptors and the cardiac uncoupling proteins 2 and 3.Iran J Basic Med Sci. 2022 Jan;25(1):126-133. doi: 10.22038/IJBMS.2022.60888.13485. Iran J Basic Med Sci. 2022. PMID: 35656450 Free PMC article.
-
Mesenchymal Stromal Cells Overexpressing Farnesoid X Receptor Exert Cardioprotective Effects Against Acute Ischemic Heart Injury by Binding Endogenous Bile Acids.Adv Sci (Weinh). 2022 Aug;9(24):e2200431. doi: 10.1002/advs.202200431. Epub 2022 Jul 3. Adv Sci (Weinh). 2022. PMID: 35780502 Free PMC article.
-
Farnesoid X Receptor Activation Protects Liver From Ischemia/Reperfusion Injury by Up-Regulating Small Heterodimer Partner in Kupffer Cells.Hepatol Commun. 2020 Feb 13;4(4):540-554. doi: 10.1002/hep4.1478. eCollection 2020 Apr. Hepatol Commun. 2020. PMID: 32258949 Free PMC article.
References
-
- Movassagh M, Foo RS. Simplified apoptotic cascades. Heart Fail Rev. 2008;13:111–119. - PubMed
-
- Choudhary R, Baker KM, Pan J. All-trans retinoic acid prevents angiotensin II- and mechanical stretch-induced reactive oxygen species generation and cardiomyocyte apoptosis. J Cell Physiol. 2008;215:172–181. - PubMed
-
- Shan P, Pu J, Yuan A, Shen L, Shen L, Chai D, He B. RXR agonists inhibit oxidative stress-induced apoptosis in H9c2 rat ventricular cells. Biochem Biophys Res Commun. 2008;375:628–633. - PubMed
