

Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis
- PMID: 22261725
- PMCID: PMC3433915
- DOI: 10.1074/mcp.O111.016717
Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis
Abstract
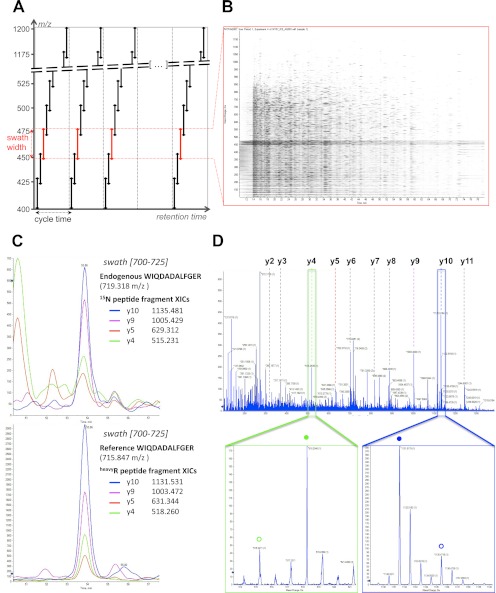
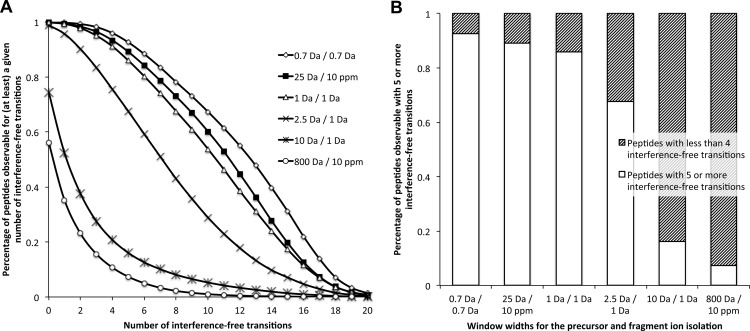
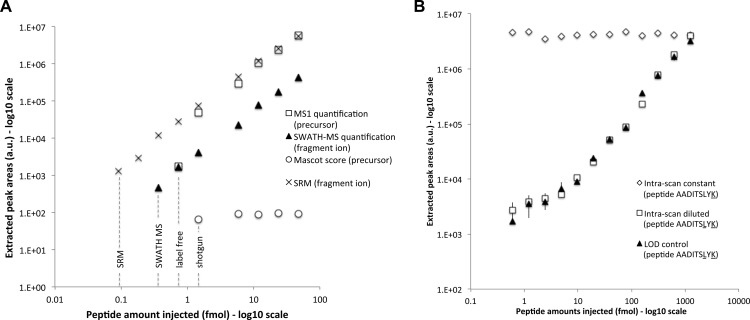
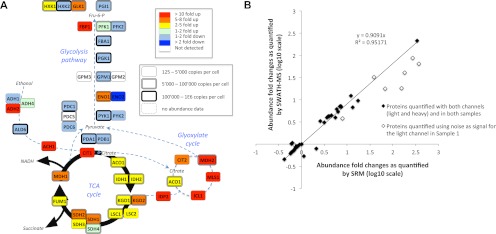
Most proteomic studies use liquid chromatography coupled to tandem mass spectrometry to identify and quantify the peptides generated by the proteolysis of a biological sample. However, with the current methods it remains challenging to rapidly, consistently, reproducibly, accurately, and sensitively detect and quantify large fractions of proteomes across multiple samples. Here we present a new strategy that systematically queries sample sets for the presence and quantity of essentially any protein of interest. It consists of using the information available in fragment ion spectral libraries to mine the complete fragment ion maps generated using a data-independent acquisition method. For this study, the data were acquired on a fast, high resolution quadrupole-quadrupole time-of-flight (TOF) instrument by repeatedly cycling through 32 consecutive 25-Da precursor isolation windows (swaths). This SWATH MS acquisition setup generates, in a single sample injection, time-resolved fragment ion spectra for all the analytes detectable within the 400-1200 m/z precursor range and the user-defined retention time window. We show that suitable combinations of fragment ions extracted from these data sets are sufficiently specific to confidently identify query peptides over a dynamic range of 4 orders of magnitude, even if the precursors of the queried peptides are not detectable in the survey scans. We also show that queried peptides are quantified with a consistency and accuracy comparable with that of selected reaction monitoring, the gold standard proteomic quantification method. Moreover, targeted data extraction enables ad libitum quantification refinement and dynamic extension of protein probing by iterative re-mining of the once-and-forever acquired data sets. This combination of unbiased, broad range precursor ion fragmentation and targeted data extraction alleviates most constraints of present proteomic methods and should be equally applicable to the comprehensive analysis of other classes of analytes, beyond proteomics.
Figures

Similar articles
-
Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification.J Proteome Res. 2012 Mar 2;11(3):1621-32. doi: 10.1021/pr2008175. Epub 2012 Feb 21. J Proteome Res. 2012. PMID: 22288382 Free PMC article.
-
Applying 'Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra' (SWATH) for systematic toxicological analysis with liquid chromatography-high-resolution tandem mass spectrometry.Anal Bioanal Chem. 2015 Jan;407(2):405-14. doi: 10.1007/s00216-014-8262-1. Epub 2014 Nov 1. Anal Bioanal Chem. 2015. PMID: 25366975
-
Optimization of Acquisition and Data-Processing Parameters for Improved Proteomic Quantification by Sequential Window Acquisition of All Theoretical Fragment Ion Mass Spectrometry.J Proteome Res. 2017 Feb 3;16(2):738-747. doi: 10.1021/acs.jproteome.6b00767. Epub 2017 Jan 3. J Proteome Res. 2017. PMID: 27995803
-
Multiplexed and data-independent tandem mass spectrometry for global proteome profiling.Mass Spectrom Rev. 2014 Nov-Dec;33(6):452-70. doi: 10.1002/mas.21400. Epub 2013 Nov 26. Mass Spectrom Rev. 2014. PMID: 24281846 Review.
-
Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial.Mol Syst Biol. 2018 Aug 13;14(8):e8126. doi: 10.15252/msb.20178126. Mol Syst Biol. 2018. PMID: 30104418 Free PMC article. Review.
Cited by
-
Potential use of EGFR-targeted molecular therapies for tumor suppressor CYLD-negative and poor prognosis oral squamous cell carcinoma with chemoresistance.Cancer Cell Int. 2022 Nov 15;22(1):358. doi: 10.1186/s12935-022-02781-x. Cancer Cell Int. 2022. PMID: 36376983 Free PMC article.
-
Data Independent Acquisition analysis in ProHits 4.0.J Proteomics. 2016 Oct 21;149:64-68. doi: 10.1016/j.jprot.2016.04.042. Epub 2016 Apr 29. J Proteomics. 2016. PMID: 27132685 Free PMC article.
-
Evolutionary Engineering Improves Tolerance for Replacement Jet Fuels in Saccharomyces cerevisiae.Appl Environ Microbiol. 2015 May 15;81(10):3316-25. doi: 10.1128/AEM.04144-14. Epub 2015 Mar 6. Appl Environ Microbiol. 2015. PMID: 25746998 Free PMC article.
-
Quantitative variability of 342 plasma proteins in a human twin population.Mol Syst Biol. 2015 Feb 4;11(1):786. doi: 10.15252/msb.20145728. Mol Syst Biol. 2015. PMID: 25652787 Free PMC article.
-
Metabolic dyshomeostasis induced by SARS-CoV-2 structural proteins reveals immunological insights into viral olfactory interactions.Front Immunol. 2022 Sep 8;13:866564. doi: 10.3389/fimmu.2022.866564. eCollection 2022. Front Immunol. 2022. PMID: 36159830 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
