

DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway
- PMID: 22237206
- PMCID: PMC3270268
- DOI: 10.1038/cddis.2011.134
DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway
Abstract
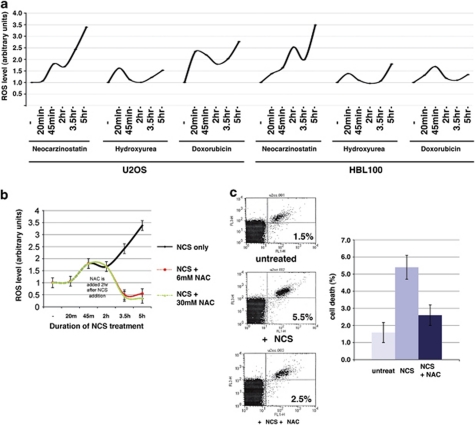
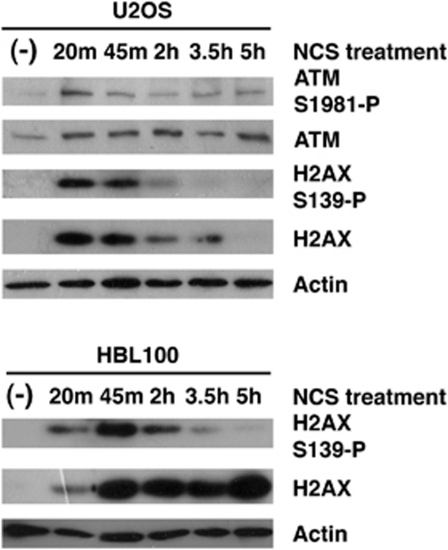
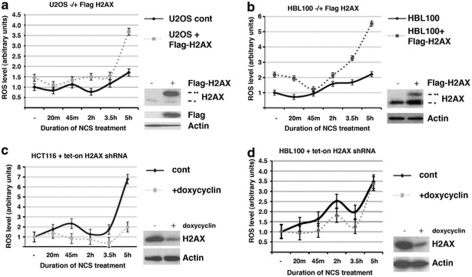
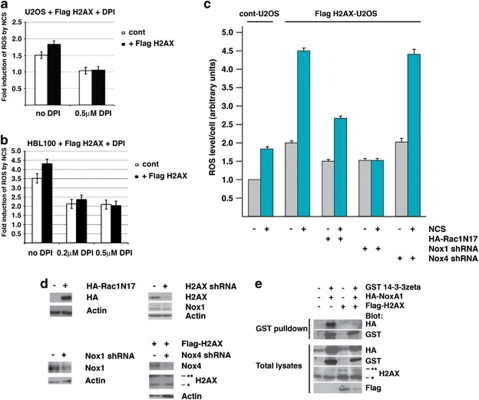
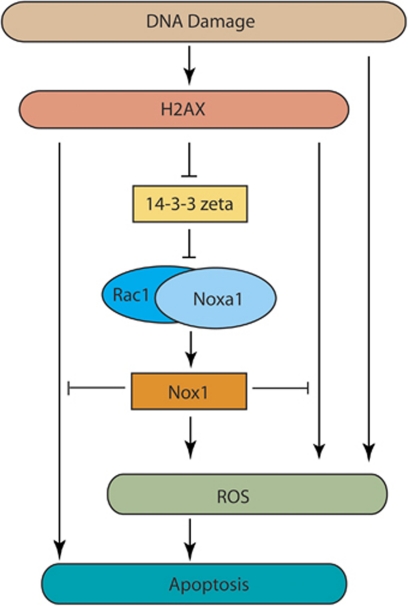
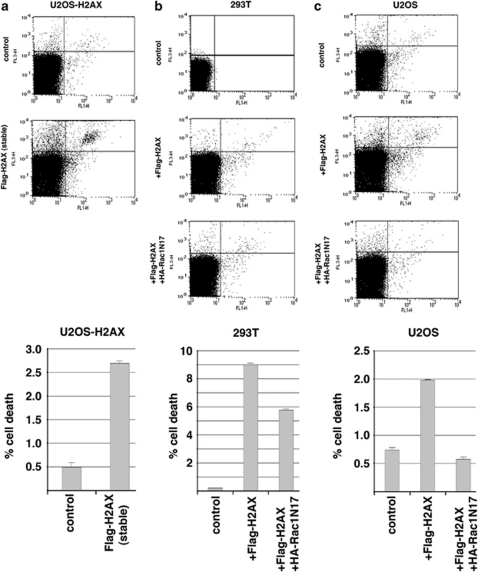
The DNA damage response (DDR) cascade and ROS (reactive oxygen species) signaling are both involved in the induction of cell death after DNA damage, but a mechanistic link between these two pathways has not been clearly elucidated. This study demonstrates that ROS induction after treatment of cells with neocarzinostatin (NCS), an ionizing radiation mimetic, is at least partly mediated by increasing histone H2AX. Increased levels of ROS and cell death induced by H2AX overexpression alone or DNA damage leading to H2AX accumulation are reduced by treating cells with the antioxidant N-Acetyl-L-Cysteine (NAC), the NADP(H) oxidase (Nox) inhibitor DPI, expression of Rac1N17, and knockdown of Nox1, but not Nox4, indicating that induction of ROS by H2AX is mediated through Nox1 and Rac1 GTPase. H2AX increases Nox1 activity partly by reducing the interaction between a Nox1 activator NOXA1 and its inhibitor 14-3-3zeta. These results point to a novel role of histone H2AX that regulates Nox1-mediated ROS generation after DNA damage.
Figures
Similar articles
-
Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases.Mol Cell Biol. 2006 Mar;26(6):2160-74. doi: 10.1128/MCB.26.6.2160-2174.2006. Mol Cell Biol. 2006. PMID: 16507994 Free PMC article.
-
Nox1-dependent reactive oxygen generation is regulated by Rac1.J Biol Chem. 2006 Jun 30;281(26):17718-26. doi: 10.1074/jbc.M512751200. Epub 2006 Apr 24. J Biol Chem. 2006. PMID: 16636067
-
Regulation of Nox1 activity via protein kinase A-mediated phosphorylation of NoxA1 and 14-3-3 binding.J Biol Chem. 2007 Nov 30;282(48):34787-800. doi: 10.1074/jbc.M704754200. Epub 2007 Oct 3. J Biol Chem. 2007. PMID: 17913709
-
Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent.Hepatology. 2012 Dec;56(6):2316-27. doi: 10.1002/hep.25938. Hepatology. 2012. PMID: 22806357 Free PMC article.
-
Organizers and activators: Cytosolic Nox proteins impacting on vascular function.Free Radic Biol Med. 2017 Aug;109:22-32. doi: 10.1016/j.freeradbiomed.2017.03.017. Epub 2017 Mar 21. Free Radic Biol Med. 2017. PMID: 28336130 Review.
Cited by
-
Limb reduction in an Esco2 cohesinopathy mouse model is mediated by p53-dependent apoptosis and vascular disruption.Nat Commun. 2024 Aug 21;15(1):7154. doi: 10.1038/s41467-024-51328-3. Nat Commun. 2024. PMID: 39168984 Free PMC article.
-
Synergistic effect of high charge and energy particle radiation and chronological age on biomarkers of oxidative stress and tissue degeneration: a ground-based study using the vertebrate laboratory model organism Oryzias latipes.PLoS One. 2014 Nov 6;9(11):e111362. doi: 10.1371/journal.pone.0111362. eCollection 2014. PLoS One. 2014. PMID: 25375139 Free PMC article.
-
Chemically different non-thermal plasmas target distinct cell death pathways.Sci Rep. 2017 Apr 4;7(1):600. doi: 10.1038/s41598-017-00689-5. Sci Rep. 2017. PMID: 28377599 Free PMC article.
-
Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy?Cells. 2024 Jun 24;13(13):1093. doi: 10.3390/cells13131093. Cells. 2024. PMID: 38994946 Free PMC article. Review.
-
Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes.Nat Commun. 2013;4:2180. doi: 10.1038/ncomms3180. Nat Commun. 2013. PMID: 23864022 Free PMC article.
References
-
- Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair. 2004;3:781–796. - PubMed
-
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. - PubMed
-
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3:959–967. - PubMed
-
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
