

TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2
- PMID: 22203995
- PMCID: PMC3268290
- DOI: 10.1073/pnas.1119296109
TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2
Erratum in
- Proc Natl Acad Sci U S A. 2012 Mar 13;109(11):4332–5
Abstract
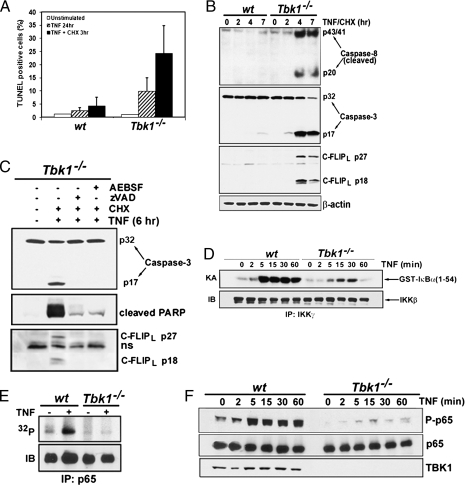
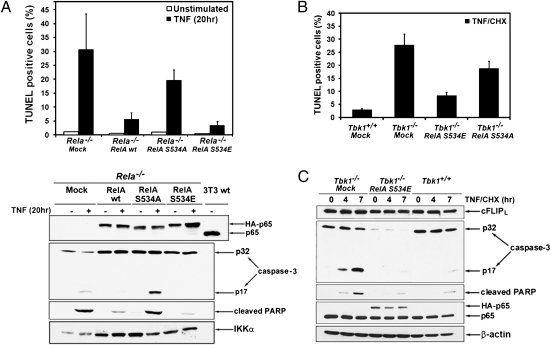
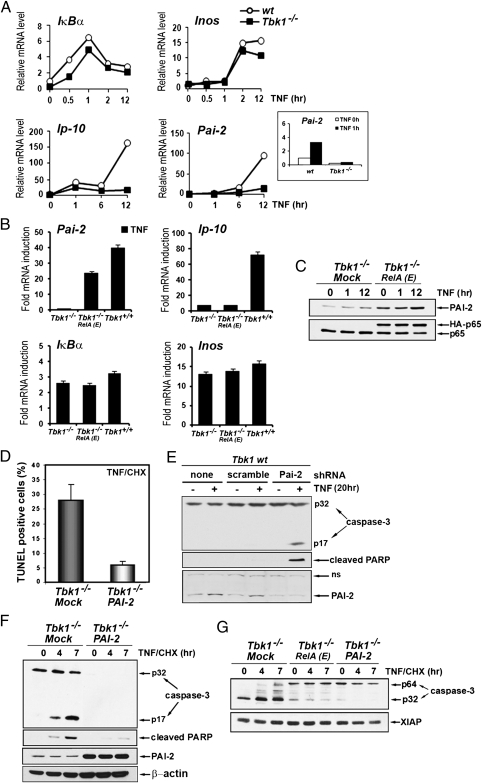
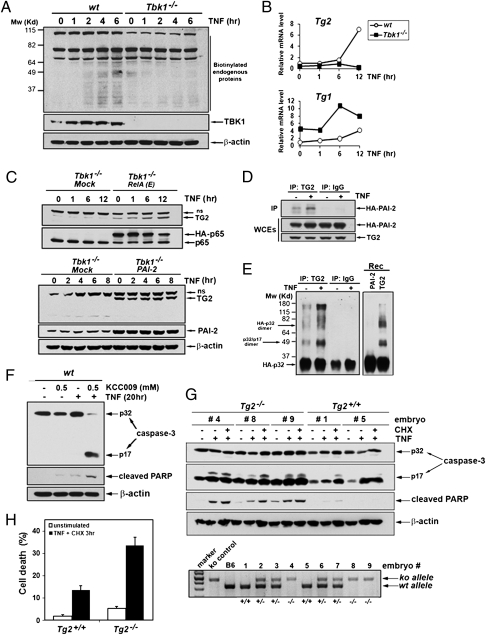
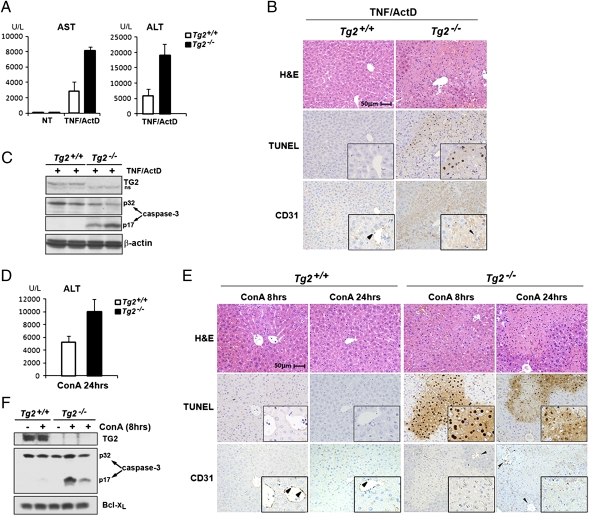
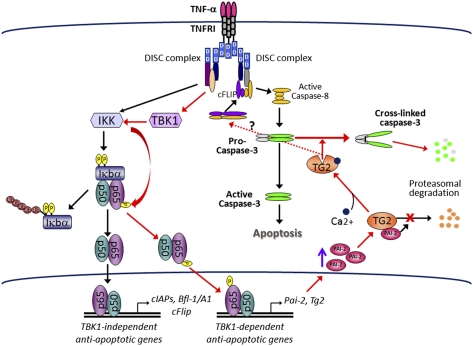
The decision between survival and death in cells exposed to TNF relies on a highly regulated equilibrium between proapoptotic and antiapoptotic factors. The TNF-activated antiapoptotic response depends on several transcription factors, including NF-κB and its RelA/p65 subunit, that are activated through phosphorylation-mediated degradation of IκB inhibitors, a process controlled by the IκB kinase complex. Genetic studies in mice have identified the IκB kinase-related kinase TANK-binding kinase 1 (TBK1; also called NAK or T2K) as an additional regulatory molecule that promotes survival downstream of TNF, but the mechanism through which TBK1 exerts its survival function has remained elusive. Here we show that TBK1 triggers an antiapoptotic response by controlling a specific RelA/p65 phosphorylation event. TBK1-induced RelA phosphorylation results in inducible expression of plasminogen activator inhibitor-2 (PAI-2), a member of the serpin family with known antiapoptotic activity. PAI-2 limits caspase-3 activation through stabilization of transglutaminase 2 (TG2), which cross-links and inactivates procaspase-3. Importantly, Tg2(-/-) mice were found to be more susceptible to apoptotic cell death in two models of TNF-dependent acute liver injury. Our results establish PAI-2 and TG2 as downstream mediators in the antiapoptotic response triggered upon TBK1 activation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription.J Biol Chem. 2004 Dec 31;279(53):55633-43. doi: 10.1074/jbc.M409825200. Epub 2004 Oct 15. J Biol Chem. 2004. PMID: 15489227
-
Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription.EMBO J. 2000 Sep 15;19(18):4976-85. doi: 10.1093/emboj/19.18.4976. EMBO J. 2000. PMID: 10990461 Free PMC article.
-
Function of polo-like kinase 3 in NF-kappaB-mediated proapoptotic response.J Biol Chem. 2005 Apr 29;280(17):16843-50. doi: 10.1074/jbc.M410119200. Epub 2005 Jan 25. J Biol Chem. 2005. PMID: 15671037
-
Regulation and function of IKK and IKK-related kinases.Sci STKE. 2006 Oct 17;2006(357):re13. doi: 10.1126/stke.3572006re13. Sci STKE. 2006. PMID: 17047224 Review.
-
Tissue transglutaminase-mediated chemoresistance in cancer cells.Drug Resist Updat. 2007 Aug-Oct;10(4-5):144-51. doi: 10.1016/j.drup.2007.06.002. Epub 2007 Jul 27. Drug Resist Updat. 2007. PMID: 17662645 Review.
Cited by
-
The P Protein of Spring Viremia of Carp Virus Negatively Regulates the Fish Interferon Response by Inhibiting the Kinase Activity of TANK-Binding Kinase 1.J Virol. 2016 Nov 14;90(23):10728-10737. doi: 10.1128/JVI.01381-16. Print 2016 Dec 1. J Virol. 2016. PMID: 27654289 Free PMC article.
-
The complexity of NF-κB signaling in inflammation and cancer.Mol Cancer. 2013 Aug 2;12:86. doi: 10.1186/1476-4598-12-86. Mol Cancer. 2013. PMID: 23915189 Free PMC article. Review.
-
SerpinB2 inhibits migration and promotes a resolution phase signature in large peritoneal macrophages.Sci Rep. 2019 Aug 27;9(1):12421. doi: 10.1038/s41598-019-48741-w. Sci Rep. 2019. PMID: 31455834 Free PMC article.
-
c-Myb Dominates TBK1-Mediated Endotoxin Tolerance in Kupffer Cells by Negatively Regulating DTX4.J Immunol Res. 2023 Mar 31;2023:5990156. doi: 10.1155/2023/5990156. eCollection 2023. J Immunol Res. 2023. PMID: 37032653 Free PMC article.
-
Leukemia cell-derived microvesicles induce T cell exhaustion via miRNA delivery.Oncoimmunology. 2018 Mar 26;7(7):e1448330. doi: 10.1080/2162402X.2018.1448330. eCollection 2018. Oncoimmunology. 2018. PMID: 29900066 Free PMC article.
References
-
- Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. - PubMed
-
- Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. - PubMed
-
- Chen G, Goeddel DV. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. - PubMed
-
- Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011;278:862–876. - PubMed
-
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
